Introduction
First identified in 1930, Erdheim-Chester disease (ECD) is an extremely rare non-Langerhans cell histiocytosis characterized by symmetric osteosclerosis of the long bones of the lower extremities (96%) due to invasion by CD68+, CD163+, Langerin-negative xanthomatous histiocytes with surrounding fibrosis [1]. Although multi-organ involvement is common, less than half of patients present with any central nervous system (CNS) involvement, and CNS-confined disease is extremely rare [1]. Based upon extrapolated data from small retrospective series, systemic therapy is the mainstay of therapy, even for patients with CNS-only disease [1]. However, some neurologic complaints may be refractory to systemic therapy [1-3]. Palliative radiation therapy (RT) is a potent tool offering improved local control and symptomatic relief for brain and spine metastases of varying histology [4,5]. Yet the role for RT, whether focal or targeted to the entire neuroaxis, remains poorly defined in patients with ECD.
Case reports have described patients presenting with a single or handful of CNS lesions, typically intracranial, without obvious systemic disease [6-8]. However, we present the case of a 38-year-old patient with ECD of the medulla complicated by respiratory failure and motor deficits, which represents, to our knowledge, the first patient with disseminated leptomeningeal disease (LMD) at diagnosis without systemic disease. This case also represents one of the first reports describing the delivery of palliative craniospinal irradiation (CSI) for this neoplasm. We review this unusual presentation of ECD to highlight the difficulties associated with accurate diagnosis and management of symptomatic, disseminated, CNS-only disease. For this report, informed consent was obtained from the patient.
Case Report
A previously healthy 38-year-old male former smoker with a 20 pack-year history presented with a 4-month history of 30-pound weight loss, 2-month history of progressive left lower extremity weakness (LLE) leading to difficulty with ambulation, and intermittent headaches. A generalized physical examination was unremarkable, but detailed neurologic examination was significant for LLE weakness with 4/5 strength with hip, knee, and ankle flexion and extension. No other neurologic deficits were noted. The headaches and motor deficits were concerning for an intracranial disease process, and the weight loss and smoking history were concerning for a metastatic neoplasm.
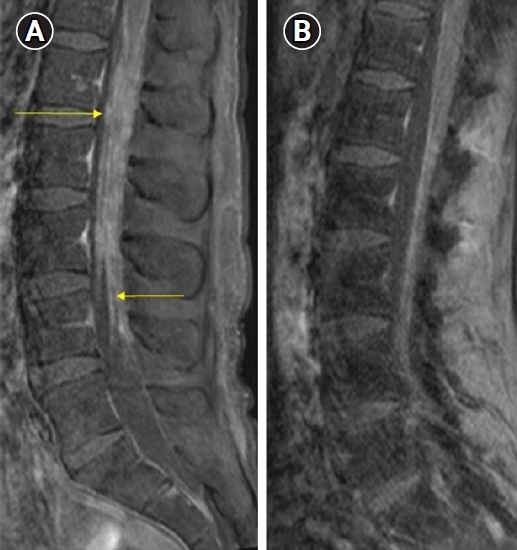
Computed tomography (CT) imaging of the chest, abdomen, and pelvis was unremarkable. However, magnetic resonance imaging (MRI) with contrast of the brain revealed diffuse, enhancing masses including most prominently a 2.4 cm × 1.4 cm lesion centered in the dorsal medulla (Fig. 1A) with mass effect causing effacement of the fourth ventricle. This mass was T1 and T2 hyperintense but not associated with edema on T2 flair sequences. A mass of the bilateral cavernous sinuses and Meckel’s caves was also noted with adjacent, intracranial LMD (Fig. 1B). Diffuse, contrast enhancing, leptomeningeal disease was also observed in the distal spinal canal and nerve roots of the cauda equina (Fig. 2A). These lesions were hypermetabolic on positron emission tomography (PET) imaging. No PET-avid disease was observed outside the CNS. Cerebrospinal fluid cytology from two separate lumbar punctures contained a few atypical cells suspicious for neoplasm but was ultimately non-diagnostic. During workup, the patient’s weakness progressed to such extent that he struggled to lift his bilateral lower extremities against gravity.
To establish a diagnosis, the patient underwent suboccipital craniectomy with C1 posterior arch resection for biopsy of the lesion of the medulla. Surgical pathology was notable for sheets of moderately pleomorphic histiocytes with prominent nucleoli, abundant eosinophilic cytoplasm, and fibrosis (Fig. 3A). CD68, a marker of histiocytic differentiation, was positive within these cells (Fig. 3B). The cells were also positive for CD163. Scattered Rosenthal fibers were also identified (Fig. 3C). Even after biopsy, the diagnosis remained unclear. The pattern of CNS-limited disease was perhaps compatible with a demyelinating condition such as multiple sclerosis or a primary tumor of the CNS such as medulloblastoma or ependymoma but incongruous with the surgical pathology. An occult primary neoplasm metastatic to the CNS such as melanoma was considered but similarly inconsistent with the histopathologic features. A pathology consult was requested from a specialized pathologist at another tertiary care center, and a diagnosis of ECD was ultimately made, as it was most consistent with the morphologic and immunophenotypic features from the surgical pathology including the sheets of pleomorphic histiocytes and CD68 positivity. Additional testing revealed that the tumor did not harbor an oncogenic BRAF mutation.
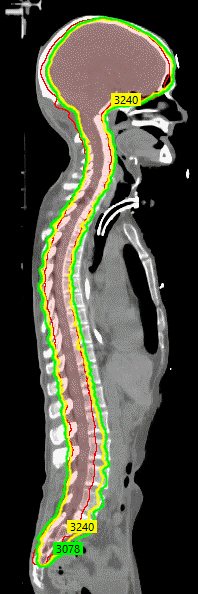
Multidisciplinary tumor board recommended that the patient receive upfront CSI rather than systemic therapy due to the extensive burden of symptomatic, diffuse, CNS-only disease. This recommendation gained added weight when the patient was admitted on the day of his first radiation treatment for acute hypoxic respiratory failure requiring intubation. Brain MRI with contrast revealed progression of the lesion in the medulla and worsening hydrocephalus. He underwent ventricular shunt placement and started RT while intubated. A dose of 3,240 cGy in 18 fractions was prescribed. The patient was simulated in the supine position without contrast with neck extended. A thermoplastic head and neck mask and knee and foot sponges were used for reproducible immobilization. The most recent MRIs of the brain and cervical, thoracic, and lumbar spine were fused to assist with target delineation. The clinical target volume consisted of the brain and entire spinal canal with 3 mm circumferential and 8–12 mm anisotropic expansions, respectively, to create a planning target volume (PTV) with margin to account for setup uncertainty. A 100% of the dose was prescribed to the PTV, and it was well covered by the 95% and 100% isodose lines (Fig. 4). Treatment was delivered using a volumetric arc therapy plan with three fields required to cover the entire neuroaxis: brain, upper spine, and lower spine. Treatment to the brain field was delivered using three arcs with 6 MV photons, while treatment to the upper and lower spine fields was delivered using two arcs with 6 MV photons. Daily cone beam CT was used to align the patient for therapy. As his clinical condition improved during his course of RT, he was extubated, underwent tracheostomy prior to discharge, and then successfully completed his prescribed course of RT as an outpatient. Treatment was well tolerated with no significant acute toxicity.
One month after CSI, surveillance MRI brain and spine demonstrated an excellent response to therapy. The symptomatic, enhancing mass in the medulla had decreased in size and displayed decreased PET-avidity. Known LMD in the brain and cauda equina were stable and significantly improved, respectively (Fig. 2B). Next, he completed three cycles of cladribine with continued disease control. Four months after RT, an oligoprogressive, contrast enhancing, left cerebellar lesion was noted on routine MRI surveillance imaging, and he successfully underwent suboccipital craniectomy for improved local control. Five months after CSI, MRI of the brain was consistent with isolated progression of the primary tumor in the medulla with increased size of the contrast enhancing disease. Again, no systemic disease was noted on MRI spine and PET restaging. Clinically, he was stable on room air and neurologic examination demonstrated significant improvement in the strength of his lower extremities (3/5 and 4/5 in the left and right lower extremities, respectively) roughly back to his 4/5 baseline at diagnosis and improved with respect to his immediate pre-CSI baseline of bilateral 2/5 strength in those extremities. No new neurologic complaints or deficits were noted. To decrease the risk of local recurrence, he underwent postoperative fractionated stereotactic radiotherapy (FSRT) to 2,500 cGy in 5 fractions to the resection bed. For improved local control, he simultaneously underwent FSRT, also to 2,500 cGy in 5 fractions, for re-irradiation of the progressive disease in the medulla. Seven months after CSI, he died secondary to cardiopulmonary arrest of unknown etiology with stable MRI imaging suggesting that it was not secondary to progressive CNS disease.
Discussion
This reports highlights, to our best knowledge, the first reported case of ECD with disseminated LMD at the time of diagnosis but without any systemic disease. This case also represents one of the first reports describing the delivery of palliative CSI for this neoplasm. ECD is an extremely rare, non-Langerhans cell histiocytosis that classically causes symmetric osteosclerosis of the long bones of the lower extremities in over 90% of cases due to organ invasion by CD68+, CD163+, Langerin-negative xanthomatous histiocytes with surrounding fibrosis [1]. A key feature frequently aiding diagnosis is that 50%–70% of patients have BRAF V600E mutated disease [1-3]. ECD primarily presents in adults 50–60 years of age with males at mildly increased risk [1]. Prognosis is highly variable, as disease course can range from indolent to rapidly progressive and/or fatal [1]. Multi-organ involvement is common, particularly cardiovascular involvement which can be seen in roughly half of patients [1]. Potential cardiac manifestations include disease of the large blood vessels, pericardium, coronary arteries, and interatrial walls [1]. Retroperitoneal fibrosis is also commonly seen [1]. Despite the risk of multi-organ involvement, only 30%–50% of patients have any CNS involvement [1-3]. CNS involvement typically manifests as disease of the hypothalamic-pituitary-adrenal axis, meninges, or brain parenchyma [1]. While CNS involvement can be an asymptomatic radiographic finding, it can also produce symptoms depending on the affected structure that can range from diabetes insipidus to focal deficits from cranial nerve, spinal cord, or brain injury [1]. MRI is the most reliable way to identify CNS disease [1]. CNS disease is a prognostic factor of unclear importance, as it has been significantly associated with inferior survival in some [3], but not all, retrospective series [1]. ECD limited to the nervous system has been described, but is incredibly rare [1]. Isolated adult and pediatric reports have documented single site ECD confined to peripheral nerves [9], cranial nerves [10], and the brain [6,7]. One case report documented a patient presenting with multiple brain lesions before eventually developing other lesions including bony disease [8]. However, our report may be the first to document ECD with disseminated LMD at presentation but without any systemic involvement. In this context, our CNS-limited, BRAF wild type case proved especially difficult to diagnose due to this unusual presentation.
Because of its rarity (less than 1,000 cases ever reported) [11], standard of care is not well defined. Evidence guiding management is mostly limited to case series and small retrospective studies [1]. Systemic therapy, although noncurative, is the mainstay of therapy, as it appears to offer a survival benefit irrespective of CNS disease burden [1]. BRAF-targeted therapy improves outcomes in some patients with oncogenic BRAF mutations [1,12], while MEK inhibitors or chemotherapeutic agents (including cladribine) are beneficial in wild type disease [1,13,14]. The role of palliative RT is well established in the management of CNS metastases [4,5]. However, the role of palliative RT for ECD in the CNS is poorly studied, and limited to case reports and small case series [8,15-20] (Table 1). Three patients are reported to have received RT to 1,600–2,000 Gy (likely conventionally fractionated) focally directed to symptomatic sites in the brainstem and spine [16,17]. These relatively low doses provided a 66% rate of durable local control [16,17], but one patient progressed within 2 months [16]. Another patient, initially believed to have multiple meningiomas, received 50.4 Gy in 28 fractions with excellent local control but progressed elsewhere in the brain outside the treated areas within a year [8]. Four patients received palliative whole brain radiation therapy (WBRT) from 1,440 to 2,400 Gy in 8 to 12 fractions [15,16,18,19]. Six months after therapy, one patient enjoyed excellent, durable intracranial control [19], but the remaining three patients experienced symptomatic, intracranial progression 2–18 months after therapy [15,16,18].
CSI for ECD has been described even less frequently. One patient with a classic presentation of ECD with bilateral lower extremity osteosclerosis received CSI to 1,600 cGy delivered over a 5-week period for symptomatic CNS disease [20]. Six months after CSI, she displayed no clinical or radiographic evidence of CNS progression [20]. This regimen differs significantly from our prescribed regimen in both dose (3,220 cGy vs. 1,600 cGy) and time elapsed during therapy (3.5 weeks with standard daily fractionation instead of 5 weeks). Our more standard dose and fractionation was justifiable given our patient’s young age, excellent prior performance status, and the need to maximize rapid but durable local control and symptomatic relief as his symptomatic brainstem mass resulted in respiratory compromise. CSI quickly aided with patient extubation, improved motor function, and was well tolerated with no observed treatment-related toxicity. Other than oligoprogression at just two sites that was amenable to local therapy, palliative CSI overall provided excellent, durable CNS disease control and symptom palliation and prevented neurologic compromise. The patient ultimately experienced a non-neurologic death without treatment-related complications. Thus, our case demonstrates that CSI may be a safe and effective palliative therapy for patients requiring improved local control and symptom palliation of disease within the CNS. The effectiveness of palliative RT in ECD is related to the somewhat radiosensitive nature of this histiocytic process; while not curative, palliative doses can at least temporarily reduce histiocytic infiltration of involved, symptomatic tissues [16].
In summary, due to its rarity, diagnosis of ECD, a rare non-Langerhans cell histiocytosis, is difficult even when it classically presents with symmetric, lower extremity osteosclerosis, multi-organ involvement, and histopathology documenting invasion by Langerin-negative histiocytes with surrounding fibrosis. CNS-only disease is extremely rare, and particularly tricky to diagnose and manage. Systemic therapy is the mainstay of therapy but can be ineffective, and refractory neurologic complaints are seen. Yet, the safety and efficacy of RT, potentially a highly effective palliative tool, remains poorly defined. We have highlighted the first case of ECD with disseminated LMD at the time of diagnosis but without any systemic disease. This case also represents one of the first reports describing the delivery of palliative CSI for this neoplasm, which provided excellent, durable CNS disease control, symptom palliation, and prevented neurologic compromise before the patient ultimately experienced a non-neurologic death. Thus, our experience suggests that in appropriately selected cases, palliative CSI may safely offer improved local control and symptomatic relief for CNS disease.