Mechanisms of radiation-induced normal tissue toxicity and implications for future clinical trials
Article information
Abstract
To summarize current knowledge regarding mechanisms of radiation-induced normal tissue injury and medical countermeasures available to reduce its severity. Advances in radiation delivery using megavoltage and intensity-modulated radiation therapy have permitted delivery of higher doses of radiation to well-defined tumor target tissues. Injury to critical normal tissues and organs, however, poses substantial risks in the curative treatment of cancers, especially when radiation is administered in combination with chemotherapy. The principal pathogenesis is initiated by depletion of tissue stem cells and progenitor cells and damage to vascular endothelial microvessels. Emerging concepts of radiation-induced normal tissue toxicity suggest that the recovery and repopulation of stromal stem cells remain chronically impaired by long-lived free radicals, reactive oxygen species, and pro-inflammatory cytokines/chemokines resulting in progressive damage after radiation exposure. Better understanding the mechanisms mediating interactions among excessive generation of reactive oxygen species, production of pro-inflammatory cytokines and activated macrophages, and role of bone marrow-derived progenitor and stem cells may provide novel insight on the pathogenesis of radiation-induced injury of tissues. Further understanding the molecular signaling pathways of cytokines and chemokines would reveal novel targets for protecting or mitigating radiation injury of tissues and organs.
Introduction
Although conventional cancer therapies (surgery, radiotherapy, chemotherapy) produce a high rate of cure for patients with early-stage disease, many cancers recur and the majority of patients with advanced cancer eventually succumb to the disease. The limitations of conventional cancer therapies do not derive from their inability to ablate tumor, but rather from limits on their ability to do so without excessively damaging adjacent normal tissue. It is this consideration that constrains the extent of surgical resection, the dose of radiation and volume to be irradiated, and the dose and combination of chemotherapeutic drugs. Improving the effectiveness of a treatment is of no clinical value if there is no significant increase in the differential response between tumor and normal tissue (i.e., the therapeutic index).
As a means of increasing the therapeutic index, several novel approaches have been made in recent years. One strategy exploits recent advances in the delivery of radiation using image-guided precision localization to permit delivery of higher doses of radiation to well-defined tumor targets. Other strategies employ novel cellular and molecular biological findings to prevent/decrease normal tissue toxicity and/or selectively enhance the intrinsic radiosensitivity of tumor tissue. Pharmacological strategies designed to reduce normal tissue damage would complement focused high dose precision radiotherapy if the drug therapies do not inadvertently protect tumor from radiation therapy. Finding ways to prevent both acute and late toxicity in normal tissue injuries is of paramount importance, since the devastation resulting from this radiation-induced toxicity can sometimes be far worse than the initial lesion that was treated. This review highlights several preclinical and early clinical manifestations of radiation toxicity of skin, gastrointestinal, lung injury, the acute and late responding tissue and brain injury, the classical late responding tissues.
Pathogenesis of Radiation-Induced Tissue Damage
Classically, normal tissue injury following high doses of radiation is thought to result from either depletion of parenchymal and/or vascular endothelial cells and attempts have been made to determine whether parenchymal or endothelial progenitors cells are the primary targets responsible for the tissue damage. One elegant radiobiological study has been carried out to address the foregoing issue using boron neutron capture therapy [1]. The method is based on the short-ranged alpha particles released from the neutron capture reaction in the stable boron isotope. Selective irradiation of the microvasculature was achieved using intraperitoneal administration of a boron compound which remains within the endothelium, since blood brain barrier (BBB) excludes the boronated compound from the brain parenchyma. The selective vascular damage from the boron neutron capture irradiation, with only a minimal dose to parenchymal glial cells, resulted in significant demyelination and white matter necrosis indicating that endothelial cell loss plays a significant role in this pathogenesis. It is, however, interesting to note that selective irradiation of vascular endothelium has no effect on the survival of murine intestinal crypt stem cells [2] and endothelial cell damage is not causative in the eventual development of the gastrointestinal syndrome [3].
More recent molecular and cellular studies suggest that dynamic secondary reactive processes in response to vascular endothelial cell and tissue stem and progenitor cell death leads to much greater cell loss, tissue damage, fibrosis, necrosis, and functional deficits. Fig. 1 illustrates the dynamic interactions involving radiation-induced death of target cells, generation of reactive oxygen species (ROS) and subsequent secondary reactive pro-inflammatory processes and innate immune responses that are believed to contribute to selective cell loss, tissue damage, and functional deficits.

Ionizing radiation causes cell death, both parenchymal and vascular, by multiple mechanisms. Historically, the direct cytotoxicity of radiation was the first identified pathway leading to tissue injury. More recently, another pathway involving inflammation has been identified. A third pathway has been studied in the last few years that implicates the innate immune response including bone marrow-derived cells (BMDC) and both M1 and M2 macrophage (MΦ) in resultant tissue damage. Arrows represent influence of one mechanism on another and suggest potential targets for interfering with the process.
Excessive Generation of ROS Following Radiation
ROS are oxygen species which transiently exist in a more reactive state than molecular oxygen. A primary ROS is superoxide, which is formed by the one-electron reduction of molecular oxygen. This reaction is catalyzed by NADPH oxidase with electrons supplied by NADPH. Superoxide may also react with nitric oxide to form another relatively reactive molecule, peroxynitrite. Fig. 2 provides a brief schematic illustration of ROS and reactive nitrogen species (RNS) production in tissues. Under normal conditions, numerous antioxidant systems defend cells against oxidative damage. However, during times of environmental stress, ROS levels increase dramatically, overwhelming antioxidants systems and resulting in significant damage to cell structures. Damaging ROS might arise from several sources including infiltrating activated leukocytes and macrophages. Further, other cells, such as fibroblasts, can be stimulated by pro-inflammatory cytokines to produce ROS. Tissue hypoxia resulting from vascular damage is another continual source of ROS generation. Several enzymes are now recognized as being potentially able to produce ROS; perhaps the most important of these is NADPH oxidase. Another source of ROS is from xanthine oxidase. The generation of these reactive molecules is part of the innate immune system and helps to rapidly clean the wound from injury, but excessive production of ROS can lead to severe tissue damage including fibrosis and even neoplastic transformation.
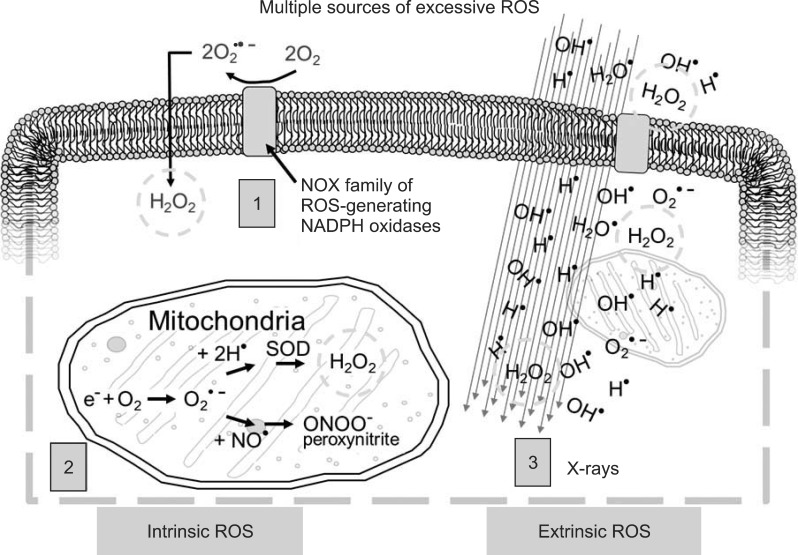
Excessive reactive oxygen species (ROS) derive from multiple sources within the cell and in response to external stimuli (1, cellular membrane ROS predominantly through NADPH oxidase; 2, mitochondrial ROS and reactive nitrogen species through cytochrome C pathway; 3, X-rays passing through the cell interact with water and other cellular molecules to produce ROS). Hydrogen peroxide (H2O2) is particularly damaging to cells (green circle) and is produced by all three mechanisms.
Most tissues and organs possess an array of defense mechanisms that interact with toxicants to obviate the deleterious effects of ROS. These include both enzymatic and non-enzymatic molecules that function as potent antioxidants or oxidant-degrading systems. ROS are cleared from the cell by enzymatic systems including superoxide dismutase (SOD), catalase, aldehyde dehydrogenase, and glutathione peroxidase, or non-enzymatic systems including antioxidants vitamins, such as alpha-tocopherol and ascorbic acid. Catalase, formed in the rough endoplasmic reticulum, is concentrated in peroxisomes located next to the mitochondria throughout the cell and reacts with hydrogen peroxide to form water and oxygen. Glutathione peroxidase reduces hydrogen peroxide by transferring the energy of the reactive peroxides to the small sulfur-containing protein, glutathione. Other known protectants include peroxiredoxin which, together with SOD, provides a powerful system against the damaging metabolites of ROS [4].
Excessive Production of Cytokines and Chemokines
Shortly after radiation exposure to the tissue and organs, a cascade of cytokines and chemokines is initiated and mediators released in the irradiated tissues perpetuate and augment the inflammatory response for long periods of time, leading to possible chronic inflammation and tissue injury (see Fig. 1). Among the numerous pro-inflammatory cytokines and chemokines that are excessively produced immediately following radiation exposure, interleukin-1 (IL-1), IL-6, tumor necrosis factor (TNF)-α, and transforming growth factor (TGF)-β are major cytokines involved in the response of skin, lung, and brain. Chemokines responsible for the recruitment of bone marrow-derived cells (BMDC) into the irradiated tissues include C-X-C chemokine ligand 12 (CXCL-12), also known as stromal cell-derived factor-1 (SDF-1) and C-X-C chemokine receptor type 4 (CXCR4).
TGF-β is a multifunctional and pleiotropic cytokine affecting many cellular processes including epithelial cell growth, mesenchymal cell proliferation, and extracellular matrix production. Irradiation, even at low doses, is one of the few exogenous factors known to induce TGF-β activation [5] and TGF-β is considered to play a central role in mediating radiation-induced tissue fibrosis (skin, lung) [6]. Elevated levels of fibrosis after thoracic and abdominopelvic radiotherapy have been correlated with the levels of TGF-β [7]. Molecular mechanisms of TGF-β signaling involve binding of TGF-β to type I/II receptor complexes leading to activation of Smad proteins. It has been shown that activation of Smad3 downstream is critically involved in the development of radiation-induced fibrosis. Smad3 knockout mice exhibit reduced radiation skin damage, reduced inflammatory infiltrates, and increased rate of epithelialization relative to the wild-type control mice [8].
TNF-α is a multifunctional cytokine involved in both acute and chronic inflammation. It is produced primarily by activated macrophages, although it can be produced by other hematopoietic and non-hematopoietic cells as well. Large amounts of TNF-α, along with IL-1, are released in response to lipopolysaccharide and other bacterial products. In addition to their pro-inflammatory effects, both TNF-α and IL-1 also stimulate the secretion of matrix metalloproteinases (MMPs). MMPs are a family of secreted proteolytic enzymes that have the capacity to degrade the basal membrane and the extracellular matrix. A local increase in TNF-α concentration in conjunction with IL-1 produce the cardinal signs of acute skin reaction, where IL-1 appears to play a critical role in the inflammatory response [9]. Several TNF-α inhibitors (monoclonal antibodies or receptor fusion proteins) have been developed and are in clinical use for autoimmune diseases, such as rheumatoid arthritis, inflammatory bowel disease, and psoriasis. Pre-clinical studies with TNF-α inhibitors have also established that they significantly mitigate radiation-induced skin injury in mice [10,65].
Vascular endothelial growth factor (VEGF) is among the first growth factors upregulated during the pathogenesis associated with late delayed effects of brain injury. The upregulation of VEGF evolves gradually following brain irradiation and typically occurs several weeks prior to the development of overt tissue pathology [11]. The cascade of events leading to white matter necrosis is initiated by a gradual depletion of vascular endothelial cells which form the BBB. The reduction in endothelial cell number is not accompanied by a substantial decrease in vascular density, rather the endothelial cell density in the existing vasculature is reduced which gradually diminishes the integrity of BBB [12]. This gives rise to vasogenic edema, inflammation, and tissue hypoxia which eventually result in the nearly simultaneous induction of HIF-1α and VEGF [13]. Reports suggesting that anti-VEGF therapy can normalize BBB function in microvessels damaged during radiosurgery in the human brain suggest that manipulating the VEGF signaling cascade may be a useful strategy for mitigating late delayed injuries resulting from brain radiation as well [14].
Role of Bone Marrow-Derived Cells in Normal Tissue Injury
As highlighted in the foregoing discussion of pathogenesis in normal tissues and organs, the well-known histological changes associated with radiation-induced tissue damage are parenchymal cell loss and vascular endothelial damage that may decrease tissue perfusion (hypoxia). In particular, one of the major causes of late effects associated with radiation injury is the initial vascular injury, which develops progressively over many months. Vascular damage can be repaired by two mechanisms: angiogenesis and vasculogenesis [15]. Angiogenesis is accomplished by the proliferation and migration of resident endothelial cells, stimulated by the concerted efforts of cytokines and growth factors. Vasculogenesis describes the de novo formation of blood vessels and is characterized by the recruitment of cells that have the ability to serve as endothelial progenitor cells. There is increasing evidence showing that local tissue damage, including radiation, causes a preferential recruitment of BMDC to the injured site [16,17]. Timing of the recruitment and homing of the mobilized BMDC into the irradiated tissue usually corresponds with the overexpression of SDF-1 and CXCR4 [16]. Three different sub-populations of BMDC are mobilized following local irradiation, i.e., mesenchymal stem cells, endothelial progenitor cells, and myelomonocytic cells. A number of recent studies have shown that myelomonocytic cells, which express the cluster of differentiation (CD), surface marker, CD11b, are the predominant BMDC that localize to the irradiated tissues including skin [18], lung [19], brain [17], and bone marrow [16]. Depending upon the local tissue microenvironment, myelomonocytic cells exhibit characteristics of macrophage lineage [20]. Although these CD11b-expressing cells do not incorporate into the blood vessels, they release angiogenic factors which stimulate vessel formation and repair in a paracrine-specific manner. On the other hand, some CD11b-expressing cells can negatively affect tissue regenerative capacity by producing a cytokine-mediated inflammatory avalanche.
Macrophages play an important role in the response to tissue injury including regulation of tissue homeostasis, coordination of the adaptive immune response, inflammation, resolution and repair. Macrophages home to the site of radiation tissue injury and may contribute to an inflammatory cytokine avalanche. Macrophage phenotypes are characterized as 'classically activated pro-inflammatory (M1)' or 'alternatively activated" anti-inflammatory (M2)' cells [20]. M1 macrophages promote inflammation, extracellular matrix destruction, and apoptosis, while M2 macrophages promote extracellular matrix construction, cell proliferation and angiogenesis. M1 macrophages tend to elicit chronic inflammation (fibrosis) and tissue injury whereas M2 macrophages tend to resolve inflammation and facilitate wound healing. Jaal et al. [21] have shown increased activity of macrophages expressing CD105 in irradiated skin tissue beginning shortly after irradiation and persisting for several weeks. We have also obtained data demonstrating increased macrophage activity (supported by the detection of elevated levels of the 160 kD glycoprotein, F4/80, expressed by activated macrophage) in the irradiated skin beginning 3 days after a single dose of 30 Gy in mice and persisting for more than 2 months thereafter. MMP9 was similarly over-expressed throughout the acute and sub-acute phases of skin injury [22]. Blocking macrophage infiltration with the use of liposomal clodronate mitigated the radiation skin injury (unpublished data) and radiation lung injury [19].
Clinical and Pathological Manifestation of Radiation Skin Injury
Acute radiation damage to the skin is manifested by early erythema and dry or moist desquamation, in part due to depopulation of the acutely responding basal epithelial cells. Such acute skin reactions usually subside over the course of time. However, some moist lesions subsequently progress into sub-acute and late phases of skin injury. Injuries to the skin may cover small areas but extend deeply into the soft tissue, even reaching underlying muscle and bone. The basal layer is repopulated through proliferation of surviving clonogenic cells; consequently, the symptoms that follow vary from dry desquamation or ulceration to necrosis depending on the radiation dose. Late effects, typically months to years post exposure, occur at doses greater than a single dose of 20-25 Gy or fractionated doses of 70 Gy or higher. The major underlying histopathological findings at the chronic stage include teleangiectasia, dense dermal fibrosis, sebaceous and sweat gland atrophy, loss of hair follicles, and with higher doses, increased melanin deposition or depigmentation and skin ulcers.
Although the specific cellular mechanisms associated with radiation-induced skin injury remain poorly understood, the pathologic effects seen in the irradiated skin are thought to result from the loss of stem and progenitor cells of the basal and dermal layers and multi-cellular interactions through a variety of inflammatory mediators leading to fibrotic processes [23]. Radiation damage to the skin is partly attributed to the reduced functioning of tissue stem cells which can no longer replace differentiated functional cells, resulting in the loss of homeostasis. Stem cell proliferation and survival is rapidly impaired within 3 days post-irradiation and a 3-fold increase in apoptosis among these cells persist for months afterward and coinciding with persistent DNA damage [24].
Clinical and Pathological Manifestation of Gastrointestinal Injury
Symptoms and signs of radiation-induced gastrointestinal (GI) injury may occur during/or after radiotherapy. The most significant symptoms of acute GI toxicity include nausea/vomiting, diarrhea, increased stool frequency, decreased food intake, fluid and electrolyte loss, and abdominal and rectal pain. Chronic GI injury develops usually 3-4 months after the completion of radiation exposure. The symptoms vary from changes in the bowel habit, diarrhea, fecal incontinence, pain and blood loss, depending on the dose and volume of radiation exposure to the intestine.
The intestinal epithelium is a highly regenerative tissue, fueled by multiple stem and progenitor cell populations at the base of the crypts of Lieberkuhn. The pathophysiology of GI injury involves dynamic interactions among the intestinal vasculature, epithelial stem cells and stromal elements including resident and infiltrating macrophages and mast cells. Microvascular injury, injury mediated by endothelial apoptosis, plays a significant and early role in the development of the acute phase of the GI injury. Vascular ischemia contributes to secondary enterocyte depletion, mucosal barrier breach, bacterial translocation, and structural damage of the intestine. Delivery of anticoagulant agents during and after high dose radiation exposure results in reduction of intestinal damage and accelerated epithelial regeneration [25]. The hallmark of the initial phase of GI injury involves the activation of ROS and their ability to stimulate transcription factors and pro-inflammatory cytokines, while the ulcerative phase is a manifestation of depopulation of the epithelial crypt stem and progenitor cells [26].
Clinical and Pathological Manifestation of Radiation Lung Injury
Radiation-induced lung injury has generally been divided into 2 sub-syndromes: pneumonitis and chronic fibrosis. Pneumonitis is characterized by acute inflammation arising within the first 3 months of radiation exposure and carries a risk for significant pulmonary complications leading to respiratory distress and organ failure. Radiation-induced fibrosis is a delayed response to radiotherapy that may develop months to years following the radiation exposure and culminates in progressive obliterative fibrosis, leading to respiratory dysfunction and decreased quality of life. Radiation pneumonitis is primarily treated with steroids. However, a retrospective study of 385 lung cancer patients with lung injury after radiation found that 16 (4.2%) died of radiation pneumonitis or steroid-induced complications, with a median survival of 45 days [27]. Current treatments for radiation-induced lung fibrosis remain ineffective in routine clinical use.
Fibrosis is caused by excessive accumulation of collagen and other extracellular matrix components. The myofibroblast is a key cell which mediates fibrosis when activated. Myofibroblasts are widely believed to differentiate from local tissue mesenchymal cells, including fibroblasts, or through an epithelial/endothelial-mesenchymal transition from epithelial and endothelial cells. Bone marrow stem cell derived fibroblast-like cells in circulation are also an important source of myofibroblasts [28].
As demonstrated with other tissues and organs being irradiated, chronic oxidative stress has been observed in irradiated lung tissue after completion of radiotherapy in lung cancer patients [29,30], as well as in preclinical rodent models of lung injury [31]. Inflammatory cells have been hypothesized to be a primary source of ROS in irradiated lung through activation of NADPH oxidase; however, non-inflammatory cells can also produce ROS. For example, myofibroblasts can produce hydrogen peroxide, which can induce epithelial cell death [32]. Chronic late effects of lung injury develop from dynamic interactions involving excessive ROS production, inflammatory cytokines and infiltration of bone marrow-derived stem and progenitors cells into the injured tissue.
Clinical and Pathological Manifestation of Radiation Brain Injury
Clinical responses of the brain to either partial or whole brain radiation may be grouped as acute, early delayed, and late delayed effects. Acute effects occur during and/or shortly after the radiation exposure and are characterized by symptoms of fatigue, dizziness, and signs of increased intracranial pressure. The acute effects are considered to be secondary to edema and disruption of the BBB. Early delayed effects occur 6-12 weeks post-irradiation and usually show reversible symptoms including generalized weakness and somnolence, in part resulting from a transient demyelination. It is, however, the late effects that may lead to severe irreversible neurological consequences. Clinical effects associated with late damage to the brain range from minor-to-severe cognitive deficits to focal or diffuse necrosis of the brain parenchyma. Late radiation necrosis may be associated with focal neurological signs, such as seizures, dysfunction of the cranial nerves, and increased intracranial pressure caused by persistent vasogenic edema resulting from BBB damage. Late radiation induced white matter necrosis can be detected using MRI where it manifests as diffuse non-specific changes in the white mater.
Since the normal brain tissue tolerance is directly related to the volume and dose of tissues being irradiated, smaller incremental exposures of the normal tissue are routinely used for clinical radiotherapy. The dose volume histogram is a useful guide in estimating the volume of normal tissues being exposed. Nevertheless, long-term functional changes following whole brain irradiation occur routinely in adults and can be more pronounced in young children, particularly when radiation is used during the developmental stages of neural tissues. Treatment-related late effects with cognitive development and learning capability as well as neuroendocrine functions are common among childhood cancer survivors [33].
Neurogenesis and Cognitive Dysfunction
Though the brain is traditionally regarded as a highly radioresistant organ, progenitor populations in the subventricular zone (SVZ) and in the subgranular zone (SGZ) of the dentate gyrus persist well into adulthood and are known to be extremely radiosensitive [34,35]. These progenitor populations represent unique sources of neurogenesis in the post-natal mammalian brain and have been consistently implicated in learning and memory function [36,37,38]. In particular, neural progenitors in the SGZ have been shown to participate in the consolidation of spatial memory, and both their proliferation and subsequent survival are increased proportional to memory demands [38]. These newly-formed neurons migrate into the adjacent dentate granule cell layer where they mature to become indistinguishable from preexisting dentate granule cell neurons [39,40]. This maturation requires several months during which these neurons are hyperexcitable and hyper-inducible with respect to activity-induced cytoskeleton associated protein (Arc), which plays a critical role in memory consolidation [41,42].
Neurogenesis within the SGZ is significantly impaired following whole brain irradiation (WBI) doses of 5 Gy and is effectively eliminated following WBI doses above 10 Gy [43]. Within this dose range, impaired neurogenesis reflects both the loss of some of the neural progenitors and a disruption of neurogenic signaling within the SGZ microenvironment required for neuronal fate determination among the surviving progenitors. The resultant loss of hippocampal neurogenesis is associated with reduced learning-induced Arc expression and, at later time-points, impaired consolidation of long-term potentiation (LTP) and behavioral learning [44,45,46,47,48].
WBI dose-dependently increases activated microglia throughout the brain which promote the development of chronic neuroinflammation [34]. Anti-inflammatory drugs can reduce microglial activation and chronic neuroinflammation within the brain, and this is sufficient to partially restore hippocampal neurogenesis along with Arc expression and the consolidation of LTP and behavioral memory [47,48,49]. Stem cell therapies have also been employed post-irradiation to replenish the progenitor population within the SGZ and have resulted in similar restoration of neurogenesis and improvements in the consolidation of behavioral memory [50].
Collectively, these data appear to establish a causal connection between radiation-induced neuroinflammation, impaired hippocampal neurogenesis, and impaired synaptic plasticity and cognitive function. However many questions remain to be addressed. There are disparities between the kinetics associated with the loss of hippocampal neurogenesis, which occurs within days of radiation exposure, and impaired cognitive function, which typically requires several months to manifest [47,48,50,51,52]. Moreover, Singer et al. [53] have recently shown that functional impairment following the ablation of hippocampal neurogenesis is transient and that within 6 weeks the loss of neurogenesis is compensated by changes in the structure and dynamics of mature neurons within the dentate gyrus. Thus, while impaired hippocampal neurogenesis may play an important role in the development of radiation-induced cognitive impairment, chronic neuroinflammation may adversely affect neuronal function by other means at later post-irradiation time points.
Therapeutic Strategies
Based on the foregoing discussion, progressive damage to normal tissues after irradiation is increasingly thought to be caused by radiation-induced long-lived free radicals, ROS and RNS, and pro-inflammatory cytokines, resulting in a deterioration of tissue and organ function. Strategies aimed at blocking effector molecules or otherwise reducing oxidative stress are attractive for preventing or mitigating radiation toxicity. Several drugs which block pro-inflammatory cytokines and ROS are currently available and in wide clinical use.
Therapeutic Strategies - Antioxidants and Anti-inflammatory Agents
Various anti-inflammatory agents including non-steroidal anti-inflammatory drugs, and cyclooxygenase-1 (COX-1) and -2 (COX-2) inhibitors have been used to mitigate skin injury. Many of these agents are administered with the intention of blocking the prostaglandin synthetase complex of enzymes that are present in normal skin. Celecoxib, a COX-2 inhibitor, produced modest mitigating effects in a pre-clinical model [54]. The strongest clinical and pre-clinical studies come from Delanian and Lefaix [55] who summarized their contributions in a review article on the current management of radiation injury (CRI). Treatments categorized as vascular directed, antioxidant and anti-inflammatory are represented by, respectively, pentoxifylline, which improves blood flow and modulates innate immunity, alpha-tocopherol (vitamin E), which is a potent antioxidant, and clodronate, which is an anti-macrophage therapy. The combination of pentoxifylline and vitamin E has been evaluated extensively by Lefaix et al. [56] and Delanian et al. [57] in a pig study and in phase II-type randomized trials. Their body of evidence indicates that this strategy is beneficial for treating CRI or for reducing fibrosis. Clodronate is the latest addition to their regimen but has not been evaluated systematically. A phase II trial of the combined therapy is underway [58].
Among the several antioxidant vitamins, vitamin E has been widely studied in various animal models of radiation injury including total body irradiation (TBI), GI injury and skin injury. Vitamin E succinate, not acetate, has been shown to protect mice from a lethal dose of TBI [59]. More recently, other isoforms of vitamin E, notably, γ-tocotrienol, have proven more effective for ameliorating the acute radiation syndrome [60]. A single dose of γ-tocotrienol administered before radiation exposure decreases radiation injury in several organ systems, including the intestine, the bone marrow and the vascular system [61]. With respect to the GI syndrome, γ-tocotrienol significantly improves intestinal crypt cell survival as well as the post-irradiation recovery of the intestinal mucosal surface area. This high level of protection against GI injury may be in part due to the high accumulating capacity within the crypt cells, mucosa and endothelial layers (30-50 fold higher).
Therapeutic Strategies - Inhibitors of Pro-inflammatory Cytokines
As discussed in the previous section, numerous therapeutic attempts have been made directly targeting TGF-β pathways, since TGF-β and its associated downstream protein, Smad, are considered to be primary determinants of the profibrotic response to radiation-induced tissue injury. Halofuginone, a plant alkaloid, is being developed for the treatment of scleroderma and it has received orphan drug designation from the US Food and Drug Administration (FDA). The drug inhibits the development of T helper cells, but it also interferes with the TGF-β signaling pathway. Xavier et al. [62] have shown the drug to be effective in mitigating radiation-induced fibrosis and leg contraction in mice, whereas similar drug treatment did not impair the efficacy of radiation treatment on tumor growth in mice. Using an inhibitor of the TGF-β receptor 1, LY-364947, others have shown markedly decreased radiation-induced tissue fibrosis [63]. Since Smad3 has been shown to be a key intracellular mediator of the effects of TGF-β, attempts have been made to mitigate radiation induced fibrosis using the topical application of Smad3 antisense oligonucleotides [64].
TNF-α is among the cytokines that stimulate the acute phase of inflammation and is produced primarily by activated macrophages. There are several FDA approved inhibitors of TNF-α available for clinical use for the treatment of rheumatoid arthritis, psoriasis and Crohn's disease, to name a few. We have recently shown that etanercept, an inhibitor of TNF-α, significantly mitigates radiation-induced skin injury in mice [65]. It is interesting to note that the timing and sequence of drug administration has a profound influence on the mitigating effects in this context. Using another pro-inflammatory cytokine inhibitor, MW-151 (Minozac), we have shown similar mitigating effects with respect to their dependence on the timing and sequence of the drug administration [22].
Therapeutic Strategies - Superoxide Dismutase Mimetics
Under normal physiological conditions, numerous cellular anti-oxidant systems exist to defend against oxidative stress and maintain the redox balance within the cell. ROS are cleared from the cell by enzymatic systems including SOD, catalase, and glutathione peroxidase, or non-enzymatic systems including antioxidants, such as alpha-tocopherol, ascorbic acid, alpha-lipoic acid, co-enzyme Q, to name a few. An early clinical study using synthetic Cu/Zn SOD has shown a promising result in the treatment of radiation induced skin fibrosis [66]. Others have used different formulations of SOD and synthetic SOD mimetics and demonstrated radio-protection in the lung, esophagus and skin injury models [67,68,69]. The use of viral vectors (e.g., adenovirus or adeno-associated virus) carrying the SOD gene have shown potential as mitigators of radiation-induced skin injury [70].
Therapeutic Strategies - Statins
Statins, HMG-CoA reductase inhibitors, block the rate limiting step of cholesterol biosynthesis. Clinically, statins are widely used to treat hypercholesterolemia and atherosclerosis. In addition to their primary mode of action, statins possess pleiotropic effects that are under active investigation. Inhibition of cellular proliferation, the restoration of endothelial activity, inhibition of platelet reactivity and an anti-oxidant potential are only a few examples of effects attributed to statins on various vascular tissues. Statins have also been shown to possess potent anti-inflammatory properties [71]. In vitro and in vivo studies demonstrate that statins reduce the generation of intracellular ROS in smooth muscle cells after stimulation of NADPH oxidase with growth factors or angiotensin [72]. There are increasing experimental data available demonstrating the neuroprotective effects of statins after ischemic and/or traumatic brain injury [73,74]. Using statins, Williams et al. [75] have shown a significant mitigation of radiation lung injury in rats. Treated animals showed a substantial reduction in both macrophage and lymphocyte populations in the irradiated lung compared to radiation alone. Statins were also shown to be effective in mitigating delayed intestinal radiation injury in rats [76,77]. We have shown that combined administration of atorvastatin with the angiotensin converting enzyme inhibitor, ramipril, mitigates the inhibitory effects of radiation on hippocampal neurogenesis after whole brain irradiation in rats [78]. Taken together, emerging data at the molecular and cellular levels are revealing an increasing number of relevant properties of statins that seem strongly connected to vascular endothelial protection and both anti-inflammatory and anti-fibrotic conditions.
Therapeutic Strategies - Angiotensin Converting Enzyme Inhibitors
Angiotensin converting enzyme (ACE) inhibitors are widely used as antihypertensive agents. ACE converts angiotensin I to angiotensin II. Angiotensin II is a potent vasopressor which acts by binding to G protein-coupled receptors, angiotensin II type 1 and type 2, respectively. Angiotensin II is a pro-inflammatory and pro-fibrogenic mediator which, in combination with other cytokines and growth factors, may participate in the development of long-term tissue and organ injury [79].
The role of ACE and its metabolites in the pathogenesis of radiation-induced tissue and organ injury was reviewed by Robbins and Dix [80]. In the mid-1980s, Ward et al. [81] first showed a modification of radiation-induced pulmonary dysfunction in rats with the use of captopril, one of the first ACE inhibitor (ACEi) used in humans. However, enthusiasm for the use of ACEi to ameliorate radiation-induced lung fibrosis was dampened when the investigators failed to demonstrate that the radioprotection was permanent (personal communication). On the other hand, Moulder et al. [82] showed that, in a rat model of radiation-induced nephropathy, the severity of both functional and histopathological injury could be reduced by captopril therapy. Importantly, the protective effect of ACEi in treating renal injury was maintained, even when therapy was non-continuous or when ACEi were used at non-hemodynamic doses [82,83,84]. Using ramipril, a lipophylic ACEi currently used in humans, Kim et al. [85] were the first to demonstrate neuroprotection against late delayed brain injury. It is significant to note that functional and histopathological protection was observed even when ramipril was administered weeks after the radiation exposure (up to 2 weeks after high single dose of radiation) [86]. More recently, they demonstrated that ramipril subtly mitigated the inhibition of neurogenesis following whole brain radiation in rats [87].
Therapeutic Strategies - Stem Cell Mobilizer
The concept that stem cells could be used for reducing normal tissue injury following radiation has been proposed for a number of years [88]. Bone marrow functions as a reservoir for progenitor cells including stromal progenitor cells, endothelial cells and hematopoietic progenitor cells. In response to tissue injury, these cells are mobilized from the bone marrow and recruited to tissues where they contribute to tissue repair and remodeling. Francois et al. [89] have shown that the administration of human mesenchymal stem cells favored healing of the cutaneous radiation syndrome in a xenogenic transplant model. However, there are currently both practical and technical complications associated with harvesting, isolating, and delivering these allogeneic cells to the desired sites. An alternative strategy for stem cell therapy is to stimulate mobilization of stem cells from bone marrow into the circulation, thereby circumventing these issues. Mobilization of bone marrow stem cells using granulocyte colony stimulating factor (G-CSF) induces repair processes, which improve function and morphology following radiation-induced damage to salivary glands in mice [90].
Plerixafor, a FDA approved drug, is a bicyclam derivative, initially developed for potential use in the treatment of HIV infection for its role in the blocking CXCR4, a chemokine receptor. Further, the compound has been found by others to be a strong inducer of mobilization of bone marrow stem cells from the bone marrow to the peripheral blood circulation. The drug produced significant mitigation in our mouse model of radiation skin injury when administered following a single high dose of radiation [91]. To our surprise, the drug was most effective at mitigating the acute skin reaction when given days after irradiation. The optimal timing coincided with the peak activity of SDF-1 expression in the irradiated tissues. It is not clear at present whether these mitigating effects of plerixafor are due to increased availability of mesenchymal or endothelial stem cells to affect repair of the irradiated tissue, or to inhibiting the influence of SDF-1 required for the homing of mobilized inflammatory myelomonocytic cells or activated macrophages to site of injury.
The recruitment of bone marrow stem cells into a site of radiation injury is a regulated process and can be modulated by multiple agents. For example, both G-CSF and plerixafor, are potent agents for progenitor cell mobilization. It has been shown that plerixafor acts on a more primitive subset of CD34+ cells than does the G-CSF [92] suggesting that a combination of both drugs might further improve mobilization of bone marrow stem cells. Indeed, clinical trials in humans have found that the combination of G-CSF plus plerixafor facilitates mobilization of bone marrow stem cells. In this regard, future work should examine the combined administration of agents for optimal mobilization and potentially improved mitigation of radiation injury.
Lastly, a question as to whether plerixafor, a CXCR4 antagonist, might protect or sensitize tumors from the effects of radiotherapy needs to be addressed. The drug has produced enhanced tumor control when combined with radiotherapy in tumor xenograft models in mice [93,94]. Furthermore, the drug has shown anti-metastatic properties when administered in multiple daily doses [95,96].
Acknowledgments
The studies were supported in part by the National Institutes of Health under awards U19-AI067734 (PI: John Moulder, Medical College of Wisconsin; Program Director Jae Ho Kim, Mitigating and treating radiation-induced CNS injury) and R21-ES019251 (PI: Jae Ho Kim), Mitigating cutaneous radiation injury with CXCR4 antagonist. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Notes
No potential conflict of interest relevant to this article was reported.