Intrathyroidal parathyroid carcinoma: a case report and literature review
Article information

Abstract
Parathyroid carcinoma is an uncommon endocrine malignancy comprising 0.5%–2% of patients with primary hyperparathyroidism. The probability of an intrathyroidal location is low (0.2%) and make preoperative suspicion and diagnosis challenging. Less than 20 cases of intrathyroidal parathyroid carcinoma have been reported. We introduce a case of intrathyroidal parathyroid carcinoma mimicking a suspicious thyroid nodule, and review the literature, with a focus on the role of adjuvant radiotherapy.
Introduction
Parathyroid carcinoma is a rare entity, accounting for 0.4% to 5% of all cases of primary hyperparathyroidism (PHPT). It is the least common endocrine malignancy, with a prevalence of 0.005% of all cancers [1]. Fritz de Quervain first described it in 1904 in a patient who presented with a non-functioning lesion. (De Quervain F, 1904) Twenty-nine years later, Sainton and Millot reported the first functioning parathyroid carcinoma. Parathyroid carcinoma arising in an ectopic location is extremely rare. (Sainton P and Millot J, 1933) Equally unusual is the presence of an intrathyroidal parathyroid gland (0.2%), which originates from inadequate migration of the parathyroid gland(s) from the third and fourth branchial arches during embryogenesis [2].
Preoperative diagnosis of an intrathyroidal parathyroid carcinoma is challenging. Due to its rarity in literature there is a lack of large-scale, multicentric series, thus natural course of parathyroid carcinoma is still unclear, and there is no universal consensus regarding management and follow-up.
Parathyroid carcinoma has a high probability of local recurrence, regional node and distant metastasis. The 5-year survival ranges from 50% to 85%. Complete surgical resection with microscopically negative margins is the primary treatment and offers the best chance of cure. Adjuvant radiation therapy is often considered as it can provide benefits in terms of local control [3].
Here, we report an unusual case of parathyroid carcinoma arising from intrathyroidal parathyroid tissue. According to the review of the related literature, less than 20 cases have been described since 1993, and this is the first case of intrathyroidal parathyroid carcinoma identified in Morocco.
Case Report
A 56-year-old female patient presented to a private practice in January 2018 with medial cervical mass evolving over a 10-month period. Family and past medical history were unremarkable. Neck examination showed enlarged thyroid gland. Thyroid ultrasound demonstrated multinodular goiter with hypoechoic irregular 35 mm nodule and prominent internal vascularity within the right thyroid lobe. These findings raised suspicion of a malignant nodule. No further imaging studies or fine needle aspiration (FNA) was performed. In February 2018, the patient underwent total thyroidectomy without lymph node dissection. Written informed consents were obtained. Pathologic examination of the thyroidectomy specimen showed an irregular gray white nodule measuring 3.5 cm × 3 cm in the right thyroid lobe with local infiltration into the surrounding thyroid tissue as well as vascular invasion compatible with intrathyroidal parathyroid carcinoma. Resection margins were positive. Thyroid nodular hyperplasia was found, with normal ectopic parathyroid in the contralateral lobe. Postoperative hypothyroidism was treated with levothyroxine at a dosage of 125 μg/day.
The patient was sent to endocrinology department for further testing. Physical examination was normal. Her body mass index was 25 kg/m2. Blood tests revealed increased serum intact parathyroid hormone (iPTH) level of 241.9 pg/mL (reference range, 15 to 65 pg/mL). Levels of serum calcium and phosphorus showed no abnormality. Thyroid, renal, and liver function tests were within normal ranges. The patient was vitamin D deficient, with serum 25-hydroxyvitamin D levels of 15.2 μg/L (reference range, 30 to 100 μg/L) and received vitamin D suppletion. Postoperative color Doppler ultrasonography displayed a 5.8 mm × 6.7 mm hypervascular hypoechoic lesion in the right thyroid bed, suggestive of residual disease. Sestamibi parathyroid scintigraphy confirmed hyperfunctioning parathyroid tissue in the right thyroid bed (Fig. 1). Chest, abdomen, and pelvis computed tomography (CT) scans ruled out metastatic disease.

Early and delayed 99mTc-methoxy-isobutyl-isonitrile (99mTc-MIBI) scintigraphy showed focal uptake in the right anterior neck, suggestive of residual disease.
Multidisciplinary board considered re-excision. The possibility of surgical treatment and risk of adverse events was discussed with the patient. She refused further surgery. According to that, radiotherapy as an alternative therapeutic approach was planned. The patient was sent to radiation oncology department for evaluation and was taken up for adjuvant treatment. Simulation was performed using the CT simulator (SOMATOM Sensation Open; Siemens, Erlangen, Germany). The patient was immobilized in supine position with a 5-point head neck and shoulder thermoplastic mask. An intravenous iodine contrast enhanced planning CT with 3-mm slice thick reconstruction was obtained. The CT DICOM images were transferred to the Monaco treatment planning system version 5.11 (Elekta, Stockholm, Sweden) for target delineation. Gross tumor volume (GTV) consisted of the macroscopic residual disease based on the postoperative ultrasonography and seen on the planning CT. A 5-mm isotropic margin was given around GTV to generate high-risk clinical target volume (CTV-HR). A low-risk clinical target volume (CTV-LR) was defined as the CTV-HR with a 5-mm margin, including the thyroid and surgical bed along with drainage lymph nodes in the paratracheal, perithyroidal areas and superior mediastinum, starting 3 mm below the skin (Fig. 2). The planning target volumes (PTVs) were created using a 5-mm expansion around the respective CTVs, except in the skin direction (Fig. 3). All the organs at risk were contoured according to the Radiation Therapy Oncology Group (RTOG) atlas for normal tissue contouring. A dose prescription of 70 Gy in 35 fractions was given to the PTV-HR, and 56 Gy to the PTV-LR.
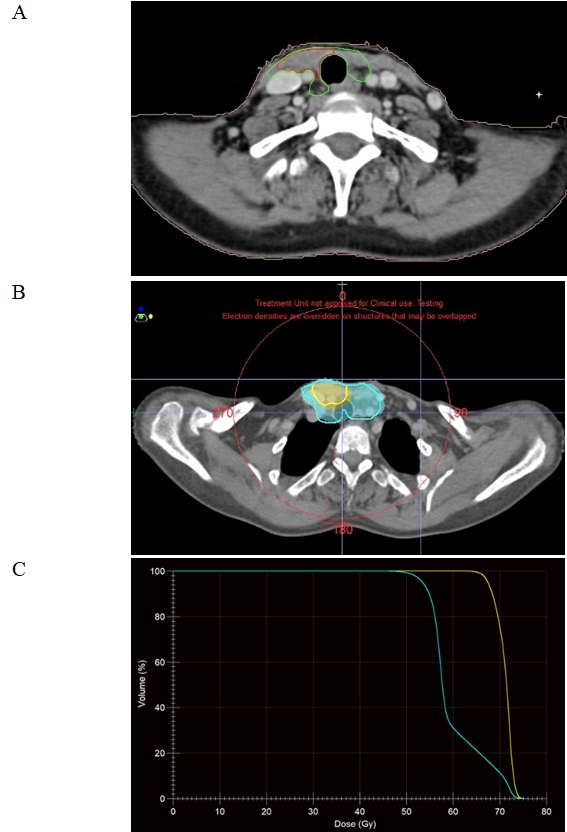
(A) Adjuvant radiation therapy was delivered via volumetric modulated arc therapy to the regions of low- and high-risk, to doses of 56 Gy (green) and 70 Gy (red), respectively. (B) Axial views of the planning computed tomography scan with 66.5 Gy (95%; in yellow), and 53.2 Gy (95%; in blue) isodose lines. (C) Cumulative dose-volume histogram for planning target volume.
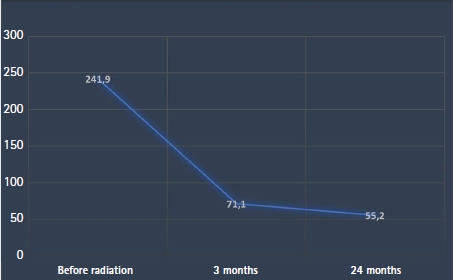
Evolution of serum parathyroid hormone levels (pg/mL) after radiation therapy.
Two-arc volumetric modulated arc therapy (VMAT) with simultaneous integrated boost (SIB) technique plan was generated using 6-MV photon beams. PTV coverage was acceptable, with a conformity index and homogeneity index of 1.06 and 0.03, respectively (Fig. 2). All the organs at risk received acceptable doses (Table 1). VMAT plan was delivered using Elekta Versa HD. Patient set-up was verified weekly by kV cone-beam CT imaging prior to treatment.
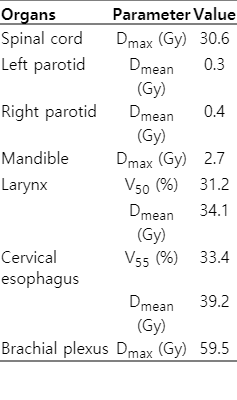
Organs at risk dosimetric parameters
Radiation dermatitis prevention consisted on local hygiene routine and use of emollient and healing creams. The patient tolerated the radiation treatment well and experienced reversible ≤grade 2 acute toxicity on skin and mucosa according to the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0.
At a 3-month follow-up visit after radiation, the serum PTH level dropped to 71.10 pg/mL and a cervical ultrasonography found an empty thyroid bed with no abnormal lymph nodes. After a period of 24-month follow-up, our patient remains asymptomatic. Her calcium and PTH levels are within normal ranges (PTH 55.2 pg/mL) and CT scans showed no signs of local or distant recurrence. The patient is still under regular follow-up till date.
Discussion
Parathyroid carcinoma is an exceedingly rare malignancy and comprises less than 2% of patients with PHPT. It affects equally both genders with a median age of 45 years [4]. The pathogenesis of parathyroid cancer remains unclear. It may occur sporadically or as a part of a genetic syndrome. It has been associated with a rare autosomal dominant inherited disorder known as hyperparathyroidism-jaw tumor syndrome (HPT-JT) due to germline mutations in the CDC73 gene that encodes parafibromin. Less frequently, it is associated with familial isolated hyperparathyroidism and multiple endocrine neoplasia type 1 and type 2A. No established risk factors have been identified. However, cases of parathyroid cancer arising in patients with prior neck radiation exposure or secondary and tertiary hyperparathyroidism from chronic renal failure have been reported [1,4].
Parathyroid glands generally lie behind the thyroid and originate from the third and fourth branchial arches [5]. Aberrant migration during embryogenesis can lead to various ectopic locations within the thymus, along the anterior surface of the carotid sheath or mediastinal. Intrathyroidal location is the most unusual site of an ectopic parathyroid gland (0.2%) [3,4].
In the literature, approximately 700 cases of parathyroid carcinoma have been reported to date and, to the best of our knowledge, less than 20 cases of intrathyroidal parathyroid carcinoma have been previously documented [6-20] (Table 2). We report a parathyroid carcinoma that was completely intrathyroidal, presenting with multinodular goiter leading to initial misdiagnosis.
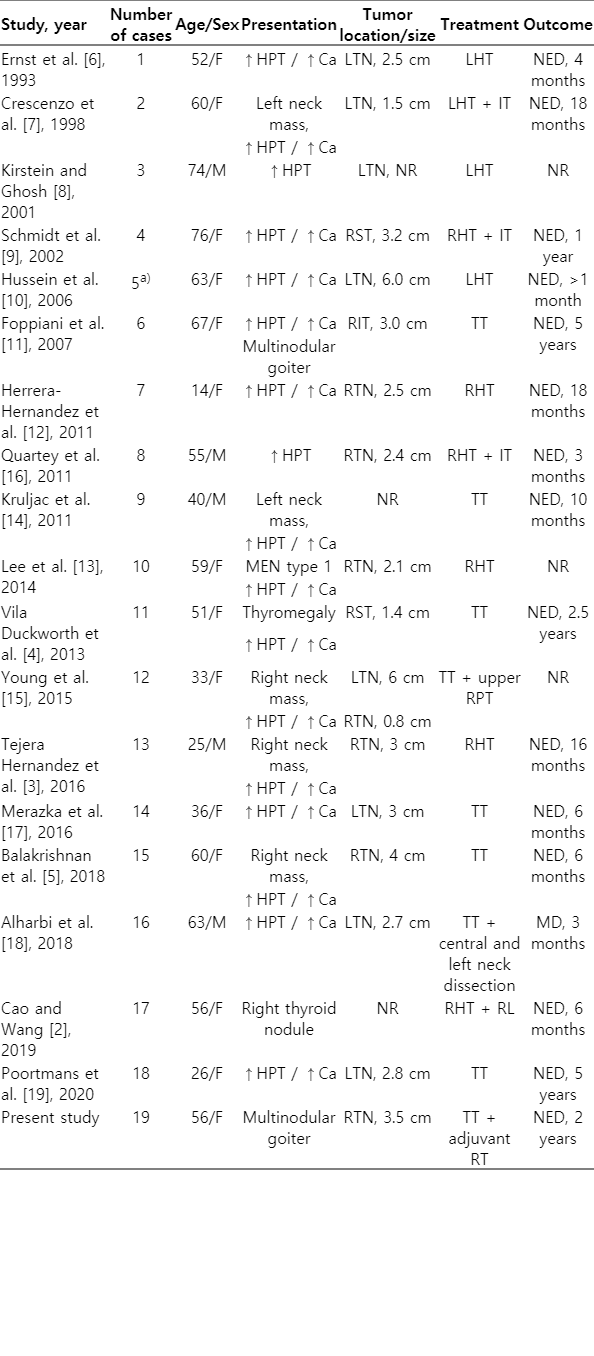
Literature review of published intrathyroidal parathyroid carcinoma cases
Patients with parathyroid cancer usually present with manifestations associated with severe hyperparathyroidism and hypercalcemia. In addition to the stigmata of hyperparathyroidism, a palpable neck mass can be present in 30%–75% of cases [1]. Asymptomatic parathyroid carcinoma is rather rare, representing approximately 2% of all. Our case is noticeable, because of the lack of symptoms on presentation except for the enlarged thyroid gland.
In absence of reliable consensual clinical diagnostic criteria, preoperative diagnosis of parathyroid carcinoma is still difficult, especially in case of infiltrative thyroid mass. The presence of multiple thyroid nodules or nodules in the vicinity of normal parathyroid glands can make perioperative and preoperative localization extremely difficult and easily missed. The clinical suspicion of severe hyperparathyroidism can be helpful. Blood sampling tests (serum calcium, iPTH, etc.) and ultrasonography of the neck are the exams of first choice when dealing with a patient with PHPT. A Sestamibi scan and MRI of the cervical area can aid in locating the affected parathyroid gland, although usually these exams cannot differentiate between adenoma and carcinoma. The same goes for FNA since there is a big overlap in cytological features of parathyroid and thyroid nodules [5]. Consequently, final diagnosis is almost always made postoperatively and is only confirmed by histological and pathological examination.
Surgery is the mainstay in the management of parathyroid carcinoma. Complete surgical resection with microscopically negative margins offers the best chance of cure and it is, therefore, recommended as a gold standard. It requires an en bloc resection of the tumor with ipsilateral thyroid lobectomy and centrocervical lymphadenectomy as well as remove of involved structures or local metastatic lymph nodes. It is of great importance that clear gross margins are obtained, and particular attention must be given to avoid capsular disruption because of the very high risk of local seeding and persistent or recurrent disease [19]. However, many patients fail to receive such treatment and experience subsequent tumor progression, such as in our case. Disease progression rates of 30%–67% have been reported [2,13]. Parathyroid carcinoma has a high recurrence rate and the potential to metastasize to regional nodes and distant sites late in its course. Patients should be closely followed over time [21]. Postoperative assay of serum calcium and PTH level is a simple and important evidence to evaluate the efficacy of treatment and to predict the recurrence of tumor as well. When complete resection is successful, this results in a 90% survival rate, if not the local recurrence rate is as high as 50%–60%. Although repeated surgical interventions have proven beneficial in palliative care, excision of progressive tumor is not consistently curative [19].
Parathyroid carcinoma is not considered to be radiosensitive, thus, no established radiotherapy protocol exists. There is no evidence for the efficacy of radiation treatment as primary therapy in local or metastatic disease. The evidence base for radiotherapy in the adjuvant setting is limited and has only been reported in small case series. The Mayo Clinic series and other studies suggested beneficial effect of adjuvant radiotherapy in preventing tumor re-growth after surgery, with reduced local recurrence and increased disease-free interval. The reported local recurrence rate without radiotherapy is 77%, whereas adjuvant radiotherapy seems to have reduced recurrence by 65% [22-29].
These results provide preliminary support that adjuvant radiotherapy may decrease the strong predilection for locoregional disease progression. However, it should be interpreted with great caution, as the studies dealing with adjuvant radiotherapy were retrospective and included a small number of patients without any comparison, thus no strong conclusion can be drawn. Therefore, the role of postoperative radiotherapy in the management of parathyroid carcinoma remains unclear. It is not recommended as a routine tool but may be of value for highly selected groups. To our knowledge, the present report is the first case of intrathyroidal parathyroid carcinoma to receive adjuvant radiation therapy. Given our patient’s refusal to undergo further surgery, adjuvant radiotherapy to the residual disease seemed essential to reduce the risk of local relapse. No recurrence occurred 2 years after treatment, with PTH level within normal ranges and without significant radiation-related toxicity.
Very few data are available in the literature regarding target volume delineation, dose prescription and fractionation or planning technique for parathyroid carcinoma. In this case, radiotherapy was planned according to the principles of the international guidelines of the Danish Head and Neck Cancer Group (DAHANCA). GTV was accurately outlined on the planning CT, based on examinations, imaging and pathology reports. Following a geometric approach, we defined a CTV-HR including the macroscopic residual tumor GTV plus a 5-mm margin and a CTV-LR adding an additional 5-mm margins. CTV-LR encompassed the operative bed, tracheoesophageal grooves, along with the elective nodal regions at risk for subclinical spread—the central nodal compartment (covered superiorly to the caudal edge of hyoid bone), the upper and lower paratracheal lymph node levels, the upper mediastinum (thoracic lymph node levels II, III, and IV covered to the level of the carina). Lateral cervical neck was omitted in a node-negative neck. These CTVs were adjusted on natural anatomic boundaries (bone, air cavities) and excluded the right carotid artery. PTVs contained corresponding CTVs with set-up margins of 5 mm. Using a SIB approach, gross disease was treated to 70 Gy in 35 daily fractions of 2 Gy, and low risk areas received a prophylactic dose of 56 Gy in 35 daily fractions of 1.6 Gy, 5 days a week over 7 weeks.
Our patient didn’t receive concomitant chemotherapy as there is no evidence supporting the efficacy of chemotherapy in parathyroid carcinoma. No standard chemotherapy regimen is available as experience is limited to case reports. A few chemotherapy drugs were used in the metastatic setting, including cyclophosphamide, 5-fluorouracil and dacarbazine as single or combination therapies, but no survival benefit was demonstrated. Denosumab, a monoclonal antibody against RANKL (receptor activator of nuclear factor kappa-Β ligand) that inhibits osteoclast maturation, function, and survival can be useful in malignant hypercalcemia treatment [27].
An interdisciplinary team approach including endocrinologists, surgeons, radiation oncologists, and pathologists must be involved to offer patients the best option for cure of this rare disease.
In conclusion, parathyroid carcinoma is a rare disease, usually presenting with clinical signs of severe hypercalcemia, but often disguising as apparently benign PHPT. It is a particularly difficult diagnosis to establish when the disease is detected outside of its typical anatomical sites. Of these, the intrathyroidal occurrence of parathyroid carcinoma can be problematic.
Clinical suspicion prior to surgery is crucial, since specific surgical procedure is the only potentially curative therapy. The combination of high levels of serum calcium, tumor size and an inverted (>1) PTH ratio are efficient in orienting towards an oncological surgical approach. The severity of this pathology is due to the severe hypercalcemia aggravating the mortality and to the risk of local recurrence and distant metastases justifying the prolonged monitoring. Adjuvant radiation therapy has been reported to effectively decrease the local relapse rate and to improve the disease-free interval, especially in high-risk patients. Whether adjuvant radiation therapy should become the standard of care in these patients remains a subject of discussion.
Notes
Conflict of Interest
No potential conflict of interest relevant to this article was reported.