Introduction
Radiotherapy (RT) is a major treatment modality for cancer patients. It is applied for approximately 50% of all cancer patients as curative or palliative treatment. RT is often used in combination with surgery, chemotherapy, or targeted therapy. Ionizing radiation delivered by RT induces DNA damage [1], which leads to tumor cell death through senescence, apoptosis, and necrosis [2]. Historically, the direct killing of tumor cells is considered the major effect of RT.
The ionizing radiation also affects lymphocytes (T cells, B cells, and natural killer [NK] cells), which are the most radiosensitive cells in the tumor microenvironment (TME) [3]. Moreover, systemic lymphopenia after localized RT has been observed in patients with solid tumors, such as high-grade glioma, lung cancer, head and neck cancer, esophageal cancer, pancreatic cancer, and cervical cancer [4-6]. Therefore, RT traditionally has been considered to have a suppressive effect on the immune system.
However, mounting evidence suggests that RT can augment immune responses against tumors. Radiation-induced DNA damage results in cytosolic DNA accumulation in tumor cells, which in turn triggers type I interferon (IFN) production via cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway [7]. Type I IFNs activate dendritic cells (DCs), thereby promoting T cell priming [8]. During cell death, danger-associated molecular patterns (DAMPs) are released, thereby activating DCs through Toll-like receptors [9]. After the phagocytosis of tumor cells, DCs present tumor antigens to T cells through major histocompatibility complex (MHC) molecules, which results in the priming and activation of T cells in the draining lymph nodes [10]. Then, tumor-reactive T cells migrate not only to the irradiated tumor sites but also to the non-irradiated sites, leading to a systemic antitumor response (termed abscopal effect) [11]. RT also induces secretion of inflammatory chemokines and cytokines that recruit immune cells to the TME, promoting antitumor responses [12].
The antitumor effect of RT can be hampered by the activation of immunosuppressive pathways. Radiation-induced DNA damage activates ataxia telangiectasia mutated (ATM)/ataxia telangiectasia and Rad3-related (ATR)/checkpoint kinase 1 (Chk1) pathway, which results in programmed death ligand-1 (PD-L1) upregulation via signal transducer and activator of transcription (STAT)/IFN regulatory factor (IRF) pathway [13]. Type I IFNs produced by DNA damage also activate STAT/IRF pathway, contributing to PD-L1 upregulation [14]. The programmed death-1 (PD-1)/PD-L1 pathway plays a key role in tumor immune escape [15]. Immune checkpoint inhibitors (ICIs) targeting PD-1/PD-L1 pathway protect T cells from anergy and apoptosis [16]. Thus, the combination of RT and ICIs could enhance antitumor responses more potently than either treatment alone. The combined effect of RT and ICIs have been evaluated and have shown promising results in preclinical studies [17-20] and clinical trials [21-25]. Furthermore, abscopal effects which seldom occur after RT alone have been increasingly reported in patients treated with the combination of RT and immunotherapy [26].
ICIs provide durable antitumor responses in various types of cancer, but the beneficial outcomes are limited to a minority of patients. The therapeutic resistance of ICIs is associated with immunosuppressive TME where MDSCs play a role as key drivers of immunosuppression [27]. MDSCs suppress antitumor responses of T and NK cells and expand regulatory T cells (Treg), promoting cancer progression [27]. Importantly, MDSC frequency is negatively correlated with therapeutic efficacy of existing anti-cancer therapies, including chemotherapy and RT as well as ICIs [28-31]. In addition, MDSCs are associated with the clinical stage, tumor burden, and overall survival [32]. Therefore, increasing efforts have been made for targeting MDSCs combined with various cancer therapies [32]. This review focuses on the role of MDSCs in RT and introduces the rationale for the combination strategies of RT and MDSC targeting to improve cancer treatment.
Myeloid-Derived Suppressor Cells
Myeloid cells are a highly heterogenous population derived from bone marrow. They include granulocytes (neutrophil, eosinophils, and basophils) and mononuclear cells (monocytes, macrophages, and DCs). The name MDSCs was first coined for myeloid cells with immunosuppressive function in 2007 [33]. These cells are phenotypically and morphologically similar to, but functionally distinct from neutrophils and monocytes. The original intent to introduce the name MDSCs was not to define a novel population of myeloid cells, but to unify different descriptions of these cells [34].
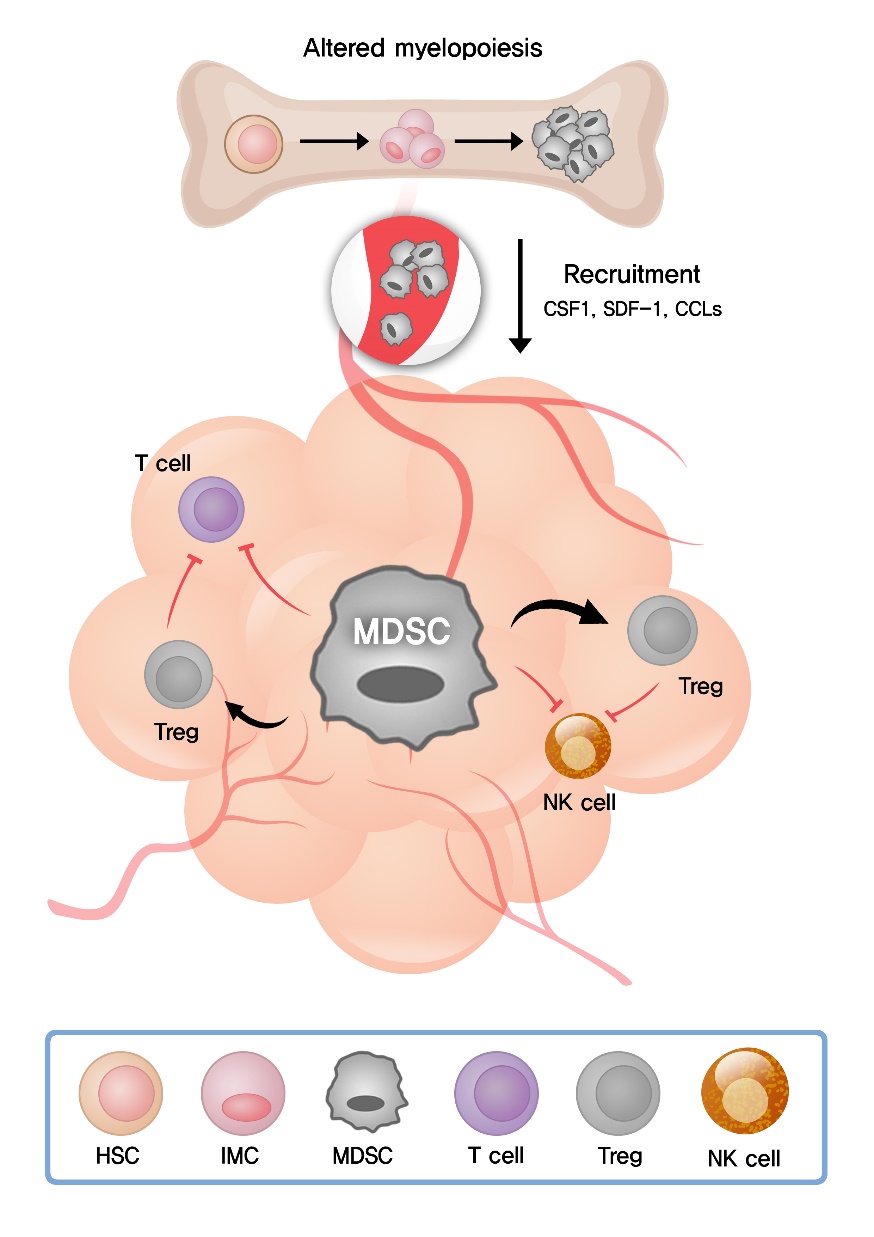
In the circulation under healthy conditions, the frequency of granulocytes and monocytes is maintained by the coordinated cytokine expression, and MDSCs are almost absent. In pathological conditions such as cancer, infection, autoimmune disease, and graft versus host disease, MDSCs are generated as result of an altered myelopoiesis [35]. Chronic inflammatory conditions such as cancer induce the production of a variety of inflammatory mediators including granulocyte colony-stimulating factor (G-CSF), macrophage colony-stimulating factor (M-CSF), granulocyte/macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-6, IL-10, IL-1β, vascular endothelial growth factor (VEGF), transforming growth factor-β (TGF-β), tumor necrosis factor-α (TNF-α), CC chemokine ligand 2 (CCL2), CCL5, and prostaglandin E2 (PGE2) [36]. These inflammatory mediators change normal myelopoiesis and skew myeloid differentiation toward MDSCs [35] (Fig. 1).
MDSCs are discriminated from other myeloid cells in which they possess potent immunosuppressive activity. Using a wide range of suppressive molecules, MDSCs suppress the functions of T and NK cells and promote the differentiation of Treg [32] (Fig. 1).
One of the main immunosuppressive molecules is arginase 1 (Arg1), which converts L-arginine into L-ornithine and urea. As a result of the enzymatic reaction, L-arginine is depleted from the TME. The lack of L-arginine causes downregulation of the T cell receptor (TCR) ζ-chain and G0/G1 phase cell cycle arrest in infiltrating T cells [37]. The induction of MDSCs in hepatitis C infection suppresses IFN-γ production of NK cells via L-arginine depletion [38].
MDSCs also overexpress indoleamine 2,3-dioxygenase (IDO), which correlates with increased infiltration of Treg in tumors and metastatic lymph nodes [39]. IDO converts L-tryptophan into N-formylkynurenine. The lack of tryptophan and production of kynurenine result in down-regulation of TCR ζ-chain in CD8+ T cells. Additionally, kynurenine produced by IDO activity induces regulatory phenotype in naïve CD4+ T cells [40].
Another important factor produced by MDSCs is suppressive reactive oxygen species (ROS). Different subsets of MDSCs employ different types of oxidative stress to regulate effector T cells. Polymorphonuclear (PMN)-MDSCs exert their function through NADPH oxidase expression and ROS generation, while monocytic (M)-MDSCs express inducible nitric oxide synthase (iNOS) and generate nitric oxide (NO). High levels of ROS induce T cell apoptosis and TCR ζ-chain downregulation. Reacting with NO, ROS nitrosylates the TCR, resulting in T cell anergy defined as a lack of responsiveness to antigen [41].
MDSCs secrete immunosuppressive cytokines such as TGF-β and IL-10 and reduce antitumor activity of effector T cells [36]. Furthermore, MDSCs exerts their immunosuppressive effects via upregulation of PD-L1. The binding of PD-L1 to PD-1 receptor on T cells leads to the anergic state in T cells [42].
In mice, MDSCs were first phenotypically identified by the expression of CD11b (a classical myeloid lineage marker) and Gr-1. The Gr-1 is a common marker of Ly6G and Ly6C molecules. MDSCs were initially defined as CD11b+Gr1+ cells, but this criterion is not sufficient to identify different subsets of MDSCs. According to the relative expression of Ly6G and Ly6C, MDSCs are classified into two subsets, PMN-MDSC (CD11b+Ly6G+Ly6Clo) and M-MDSC (CD11b+Ly6G-Ly6Chigh). Human MDSCs are generally identified based on the expression of the myeloid marker CD11b and low or absent HLA-DR. The equivalent to PMN-MDSC is further defined as CD14–CD15+, and M-MDSC is defined as CD14+CD15– [34].
However, theses phenotypic evaluations cannot discriminate PMN-MDSCs from neutrophil, and M-MDSCs from monocytes. Presently, there are no unique phenotypic marker for MDSCs. Therefore, an algorithm was proposed to identify cells as MDSCs. First, pathological conditions such as chronic inflammation and cancer should be associated with an expansion of cells with an MDSC phenotype. Then, isolated cells with MDSC phenotype must have immune suppressive activity [34].
MDSC Targeting for Cancer Treatments
It is important to prevent T cell inhibition to boost the existing antitumor responses as proven by the immune checkpoint targeting, but it is also important to regulate the TME for effective cancer treatment. In recent years, the accumulation of MDSCs have been highlighted as a major contributing factor in the immunosuppressive TME. The efficacy of MDSC inhibition have been evaluated in numerous preclinical and clinical studies [32]. There exist different approaches to target MDSCs: inhibition of MDSC accumulation, blocking of MDSC recruitment, and inhibition of MDSC function [32] (Table 1). Recently, MDSC inhibition in combination with immunotherapy have shown promising results in humans [43]. Clinical trials targeting MDSCs are summarized in Table 2.
1. Inhibition of MDSC accumulation
The frequency of MDSCs can be reduced by all-trans retinoic acid (ATRA), an active metabolite of vitamin A. ATRA has been successfully used in the treatment of acute promyelocytic leukemia where it terminally differentiates immature myeloid cells into mature myeloid cells, resulting in leukemic cell death [44]. This concept of differentiation therapy provides a rationale for the use of ATRA in reducing MDSC accumulation. ATRA induced the differentiation MDSCs into DCs and macrophages, and thereby improved T cell function in mouse and human samples [45,46]. The antitumor effects of ATRA have been extensively studied in numerous studies in the past decades, but MDSCs were not almost evaluated in these studies [47]. In some clinical studies, ATRA has been reported to reduce the frequencies of circulating MDSCs [43,48,49]. ATRA treatment improved the immune response to cancer vaccine [48] and antigen-specific T-cell response [49].
Chemotherapeutic agents such as gemcitabine and 5-fluorouracil have been shown to reduce the number of MDSCs and to enhance antitumor immunity in mouse tumor models [50,51]. These agents had no significant effect on the frequencies of T cells and NK cells [50,51]. Similar to the preclinical observations, gemcitabine reduced MDSC frequency in the peripheral blood of patients with pancreatic cancer [52]. In a recent report, a liver X receptor (LXR) beta agonist, RGX-104 induced MDSC apoptosis in the periphery and TME, leading to enhanced T cell-mediated antitumor immunity in various mouse tumor models [53].
Sunitinib is a tyrosine kinase inhibitor acting toward VEGF receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), KIT and fetal liver tyrosine kinase receptor 3 (FLT3). It was approved for the treatment of patients with advanced renal cell carcinoma (RCC) and imatinib-resistant gastrointestinal stromal tumor (GIST) in 2006 [54]. Since VEGF was implicated in the accumulation of MDSCs, the effect of sunitinib on MDSCs was evaluated [55]. The elevated levels of circulating MDSCs were decreased after sunitinib treatment in patients with RCC [56] and oligometastases [28].
2. Blocking of MDSC recruitment
MDSC recruitment to the tumor is essential process for their immunosuppressive function. This process is mediated by various chemokines secreted in the TME. MDSCs express chemokine receptor CCR2, CCR5, and CXCR2, which mobilize them to the blood and the tumor sites.
CCR2 interacts with its ligands CCL2 and CCL5, which is required for the recruitment of M-MDSCs [57]. In a phase Ib clinical trial, a CCR2 antagonist (PF-04136309) was tested in combination with nab-paclitaxel plus gemcitabine in patients with pancreatic ductal adenocarcinoma. However, the results showed that PF-04136309 did not improve the efficacy compared to nab-paclitaxel plus gemcitabine [58].
CCR5 interacts with its ligand CCL3, CCL4 and CCL5, and the CCR5-CCL5 axis is required for the mobilization of PMN-MDSCs. Targeting CCR5-CCL5 axis has been reported to block MDSC recruitment and prevent tumor growth in preclinical studies [59,60]. Intriguingly, individual who carry CCR5 deletion mutation (CCR5Δ32) are physiologically normal, whereas tumor cells overexpress CCR5. With plausible rationale for CCR5 targeting, increasing number of studies have focused on targeting CCR5 in combination with immunotherapy [61].
3. Inhibition of MDSC function
Phosphodiesterase-5 (PDE-5) inhibitors are widely used in the treatment of erectile dysfunction and pulmonary hypertension. It was reported that PDE5 inhibitors induced antitumor immune responses and substantially delayed tumor progression. PDE5 inhibitor sildenafil downregulated Arg1 and iNOS expression, and inhibited the suppressive function of MDSCs in mouse tumor models [64,65]. Recent clinical trials showed that PDE5 inhibitor tadalafil inhibited MDSC function and promoted antitumor immunity in patients with head and neck squamous cell carcinoma [66,67].
Histone deacetylase (HDAC) inhibitors have been reported to have potent immunomodulatory activity in mouse tumor models and cancer patients [68]. A class I HDAC inhibitor entinostat combined with ICIs has been evaluated in mouse tumor models [69,70]. Entinostat reduced Arg1 and iNOS expression and inhibited the immunosuppressive function of MDSCs, resulting in enhanced responsiveness to ICIs [69,70].
As described above, IDO is a key enzyme required for immunosuppressive activity of MDSCs. IDO inhibition reversed tumor-associated immunosuppression and showed antitumor effect in mouse tumor models [71]. However, clinical trials with IDO-1 monotherapy have not produced satisfactory results as observed in preclinical studies. Consequently, clinical trials have been redesigned to test IDO inhibitors in combination with other therapies, such as ICIs, chemotherapy, and radiotherapy [72].
MDSC Targeting for RT
RT has two opposite effects on MDSCs, dependent on dose-fractionation schemes and tumor models. Although it seems difficult to draw a consistent conclusion from recent studies, some patterns emerge, that is, conventional fractionated radiotherapy (CFRT) increases MDSCs while ablative hypofractionated radiotherapy (ABHRT) decreases MDSCs [73]. However, regardless of RT scheme, targeting of MDSCs has been shown to increase the antitumor effect of RT in several studies.
Radiation induced colony stimulating factor 1 (CSF1) expression through ABL1-dependent transcription. In response to CSF1, MDSCs were recruited to tumor sites and expanded systematically in the tumor, spleen, lymph nodes, and peripheral blood. CSF1/CSF1 receptor (CSF1R) blockade inhibited MDSC infiltration and tumor growth after irradiation (3 Gy × 5). In accordance with mouse studies, serum CSF1 was elevated in prostate cancer patients after RT [74]. As mentioned above, several chemokines and their receptors promote MDSC recruitment into the TME. Stromal cell-derived factor-1 (SDF-1) is a chemokine up-regulated in tumor tissues after radiation [75,76]. SDF-1 receptor CXCR4 is expressed on immunosuppressive cells including Treg and MDSCs, which are attracted by SDF-1 produced within the tumor [77]. CXCR4 antagonist AMD3100 was shown to prevent tumor regrowth when combined with radiation [75,76]. These studies suggest that CD11b+F4/80+ myeloid cells are associated with tumor regrowth after radiation. However, M-MDSCs also express F4/80 marker and distinct phenotypic markers for M-MDSCs were not used in these studies. Therefore, it cannot be ruled out that CD11b+F4/80+ myeloid cells may be attributed to M-MDSCs. Radiation-induced STING activation also contributes to MDSC infiltration. This activation caused tumor cells to produce type I IFN which, in turn induced expression of CCL2, CCL7, and CCL12, chemoattractants for MDSCs. Chemokine receptor CCR2 knockout blocked MDSC accumulation and enhanced tumor regression following radiation (20 Gy) in MC38 and LLC tumor models. Treatment with anti-CCR2 antibody also enhanced antitumor effects of radiation [78]. More recently, it was reported that radiation (12 Gy × 3) increased infiltration of MDSCs in tumors, which was suppressed by IDO1 inhibitor. Radiation combined with IDO1 inhibitor enhanced tumor growth inhibition in LLC tumor model [79].
On the other hand, several recent studies have shown that high-dose irradiation decreases MDSC frequencies. A single high-dose irradiation (30 Gy) induced complete remissions, which was related to an increased CD8+ T cell infiltration and a reduced MDSC infiltration into the TME in CT26 and MC38 tumor models [80]. In hepatocellular carcinoma (HCC) patients, the frequency of MDSCs after RT was significantly decreased and inversely correlated with overall survival. These results suggest that patients with a high frequency of MDSCs should be monitored closely and the inhibition of MDSCs may improve treatment outcomes after RT [31]. In mouse studies, ABHRT inhibited MDSC recruitment into tumors and significantly inhibited the tumor growth compared with CFRT. VEGF expression, which mediated MDSC recruitment, was relatively lower after ABHRT than CFRT. VEGFR2 blocking antibody plus CFRT reduced infiltrating MDSCs in tumors and inhibited tumor growth more efficiently than CFRT alone [81]. These results indicate that via blockade of MDSC recruitment, the therapeutic efficacy of ABHRT could be achieved and the effect of CFRT enhanced. As described above, receptor tyrosine kinase inhibitor sunitinib inhibited MDSC accumulation, and thereby restored antitumor immunity. Concurrent sunitinib and stereotactic body radiotherapy (SBRT) reversed MDSC-mediated immunosuppression and resulted in favorable clinical outcomes in patients with oligometastases [82].
When combined with ICI treatment, RT have shown improved efficacy in preclinical studies [17-20] and clinical trials [21-25]. In TUBO and MC38 tumor models, high levels of radiation (12 Gy for TUBO and 20 Gy for MC38) combined with PD-L1 blockade synergistically amplified the antitumor effect, which was correlated with a reduction of MDSCs mediated by tumor infiltrating CD8+ T cells [17]. Similarly, in LLC tumor model, radiation (6 Gy in 3 fractions) and anti- PD-L1 antibody effectively inhibited tumor growth compared to either therapy alone [19]. A recent clinical study evaluated whether SBRT could enhance the effect of ICI treatment in patients with advanced NSCLC. Interestingly, PD-L1-negative patients had the largest benefit of improved overall survival and progression-free survival in the combined treatment of RT [24]. These results suggest that RT may convert the immunosuppressive TME to a more ICI-responsive one. One more important finding is no increase in treatment-related toxic effect.
The therapeutic effect of anti-PD-L1 antibody was initially assumed to result from blockade of PD-L1 expressed on the tumor cell itself. However, several recent studies highlighted the essential roles of PD-L1 expressed on host myeloid cells. These studies suggest that PD-L1 on DCs, macrophages and MDSCs rather than tumor cells is the relevant mechanistic target for PD-1/PD-L1 inhibitors [83,84].
For the combined treatment with RT, numerous studies so far have focused on ICIs, which target PD-1/PD-L1 interactions. The available data provide evidence that the therapeutic efficacy of RT could be enhanced when combined with MDSC targeting therapy.
Conclusion
RT-mediated immune responses support both tumor immune recognition and tumor immune evasion. Thus, combining RT and immunotherapy can be a rational strategy to improve cancer treatment. Numerous studies have shown promising results in the combination of RT and immunotherapy. However, translational research behind this approach is still needed to maximize the therapeutic efficacy and increase the response rate in cancer patients. In the light of therapeutic resistance, recent studies highlight the immunosuppressive TME which promote tumor immune evasion. MDSCs are increasingly recognized as important contributors to immunosuppression in the TME and are also closely associated with resistance to RT. Therefore, MDSC targeting could be a successful complementary strategy for RT, and also RT combined with other therapies. Further work is needed to identify specific markers for MDSCs, which would enable the development of methods to selectively target these cells. Improving specificity in targeting MDSCs could help to find the strategy to maximize the efficacy of antitumor therapies.