Introduction
The concept of a radiation therapeutic ratio for cancer treatment has evolved over the years and generally centers around efforts to maximize the radiation response of cancer cells to achieve local control, while minimizing the potential for radiation-induced acute and late morbidity on normal tissues [1,2]. Both parameters have sigmoidal dose-response curves. Thus, a favorable outcome in dose-response curve is the response of tumor tissue is greater than that of normal tissue to the same dose, meaning that the therapy is effective to tumors, whereas overlapping response of two tissues is highly likely to cause serious consequences to normal tissues and ineffective treatment to tumors. Classically, fractionated radiotherapy (RT) is favored over single large doses, mainly because fractionated RT reduces the potential for late normal tissue toxicity. However, the therapeutic ratio becomes marginal with fractionated RT, especially with large solid tumors. Consequently, three distinct strategies have been evolved to improve its utility. One strategy exploits recent advances in the delivery of radiation using image guided precision localization to permit the delivery of higher doses of radiation to well-defined targets. A second strategy exploits advances in targeting radiation using radiosurgery and/or intensity modulated radiotherapy to minimize exposure and toxicity in the surrounding normal tissues. Both of these strategies are aimed at delivering much higher radiation doses to the tumor tissue, nevertheless the normal tissue and organ tolerance remains the limiting factor. A third strategy involves the application of recent cellular and molecular findings to pharmacologically prevent/decrease normal tissue toxicity or enhance the intrinsic radiosensitivity of tumor tissue. This review highlights pre-clinical and early clinical data derived from the biological targeted cancer therapies that are translatable for future clinical trials.
Targeted Cancer Therapy and Radiotherapy
The interaction between targeted therapies and RT is of clinical importance for at least two reasons. Targeted cancer therapies are often combined with conventional approaches such as RT, so the effect of targeted therapies on radiation effectiveness and vice-versa is important. Additionally, targeted therapies act on specific molecular targets that are associated with cancer, whereas radiation (and most standard chemotherapies) acts on all rapidly dividing normal and cancerous cells. Consequently, the effects of targeted therapies are often cytostatic, whereas radiation (and conventional chemotherapy agents) generally kill rapidly dividing cancer cells. Specific inhibitors have been developed targeting the signal transduction pathways. Among them, three pathways are of particular interest when combined with radiation. The PI3K/mTOR pathway has been extensively studied in conjunction with radiation [3,4]. Relatedly, the metabolic state of cancers and targeted metabolic reprogramming critically affect radiotherapy effectiveness [5]. Another pathway of interest in the cancer radiobiology is DNA damage response and checkpoint regulators of the cell cycle [6].
1. PI3K/mTOR and radiotherapy
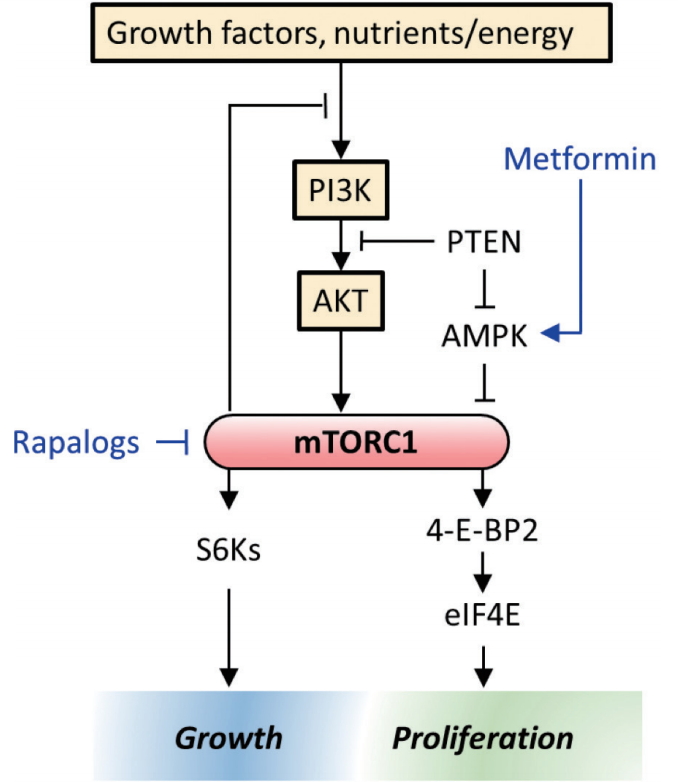
The PI3K/AKT/mTOR pathway is an intracellular signaling pathway important in regulating the cell cycle. It is directly related to cellular quiescence, proliferation, cancer, and longevity. The protein complex mTORC1 is a convergence point for multiple signaling pathways that control cell growth and proliferation. mTOR is a nutrient-sensing enzyme that regulates cell growth in nearly all eukaryotic species. Since the PI3K/mTOR pathway is frequently activated or dysregulated in many tumors, several classes of agents targeting this pathway are in under clinical development (Fig. 1). Currently, rapamycin analogs (rapalogs) are in clinical use as single or combination agents (e.g., renal, breast, pancreatic, gastrointestinal and lung cancers).
Since PI3K/mTOR pathways are altered in many human cancers and the increased activation of these signaling pathways have been implicated in the tumor radioresistance, numerous studies have been carried out to determine the extent of radiation modification using in vitro and in vivo cancer models. Early studies showed enhanced radiation damage of tumor vasculature when radiation was combined with mTOR inhibitor (e.g., everolimus) in an in vivo mouse tumor model but no in vitro radiosensitization of cancer cells grown in cell culture, hence, it was concluded that mTOR inhibitors exert their enhanced radiation response by inhibiting angiogenesis [7,8]. Subsequent reports have shown that mTOR inhibitors enhance the radiation response of tumor cells both in vitro and in vivo regardless of the timing and schedule of mTOR inhibition [9-12]. More recently, Hayman et al. [13,14] further showed that the inhibition of mTOR reduces eIF4E, a key factor in the translation which initiates the formation of the eIF4F cap complex required for cap-dependent translation. Elevated eIF4E is also correlated with both chemo and radioresistance and selectively silencing eIF4E was shown to enhance the radiosensitivity of cancer cell lines but not normal cells.
At the cellular level, other investigators showed that mTOR inhibitors (e.g., temsirolimus) could overcome the radioresistance of hypoxic tumors, since the upregulation of mTOR regulates the hypoxia-inducible factor-1α (HIF-1α) pathway [15]. Indeed, mTOR inhibition reduces oxygen consumption through inhibition of the mitochondrial respiratory complex I [16]. Our own data show that increased radiosensitivity of tumor cells is in part related to increased ROS production through a feedback loop involving AKT suppression and upregulation of AMPK (manuscript in preparation).
What is clear is that the mechanisms by which mTOR inhibitor enhances tumor radiosensitivity are multi-factorial. Currently, several clinical phase II studies are underway in which everolimus is administered in combination with radiation therapy for the treatment of lung and brain tumors.
2. Metabolic reprogramming and radiotherapy
In recent years, metformin, an oral anti-diabetic biguanide medication for type II diabetes, has attracted interest as a cancer therapeutics. The widespread use of metformin as an anti-diabetic drug, its expected safety profile, and encouraging pre-clinical data led to the several clinical trials evaluating the anti-cancer properties of metformin in combination with chemotherapeutic drugs and fractionated radiotherapy.
Though metformin has been in use for decades to reduce blood sugar levels, the exact mechanisms by which it exerts this anti-diabetic effect is not completely understood. It is widely accepted that metformin decreases hepatic gluconeogenesis and lowers insulin levels, with strong experimental evidence suggesting that it decreases ATP production by acting on mitochondrial respiratory complex I, ultimately activating AMP kinase with secondary inhibition of protein synthesis [17]. This pathway is known to affect the downstream PTEN/PI3K/AKT/mTOR system which plays a pivotal role in cancer’s unregulated growth (Fig. 1).
Mechanistic studies to date have demonstrated that metformin enhances the therapeutic gain of RT in multiple ways. Some researchers [18,19] have shown that metformin enhances the radiosensitivity of MCF-7 human breast cancer cells and lung cancer cells in both in vitro and in vivo models. They further showed that radiation and metformin together activate AMPK leading to inactivation of mTOR and suppression of its downstream effectors such as S6K1 and 4EBP1. Suppression of the 4EBP1 protein reduces eIF4E, enhances the radiosensitivity of several human cancer cells (see section in PI3K/mTOR and Radiotherapy). Others suggest that metformin reduces tumor hypoxia, presumably through the inhibition of mTOR pathway and that it may be beneficial if administered prior to single large dose of radiation [20]. In addition to its inhibition of HIF-1α via the mTOR pathway, metformin is an inhibitor of complex I in the mitochondrial respiratory chain so that the utilization of molecular oxygen is reduced, thereby making the excess oxygen available to the tumor microenvironment.
Data suggest that the mechanism of radiosensitization by metformin may derive not only from re-oxygenation of the hypoxic tumor but from other cellular mechanism as well, especially when metformin is combined with high dose per fraction (more than 8–10 Gy per fraction) radiation. High doses of irradiation is known to shut down the tumor vasculature and the subsequent radiation-induced hypoxia and low glucose would preferentially favor tumor necrosis [21]. These data provide a pre-clinical rationale of combining radiosurgery and single high dose metformin in the clinical settings. Currently, a randomized phase II trial of concurrent chemoradiotherapy +/- metformin in locally advanced NSCLC is underway [22].
Arsenic trioxide (ATO) is a US Food and Drug Administration approved anti-tumor drug (e.g., acute promyelocytic leukemia), that inhibits the mitochondrial complex III activity in the electron transport chain. Similar to results with combined metformin and radiation, a strong synergistic action of ATO has been shown when the radiation is delivered shortly after the drug administration. Mitochondrial inhibitors such as metformin or ATO, have a marginal cytotoxic effect when administered alone, whereas combining with these inhibitors with radiation has a dramatic cytotoxic effect [23,24].
3. Cell cycle checkpoint inhibitors and radiotherapy
The integrity of DNA replication and repair during cell proliferation is normally tightly controlled by several key cell cycle checkpoint proteins, including cyclin-dependent kinase (CDK), checkpoint kinase, and WEE1 kinase (WEE1). It is well known that ionizing radiation induces both doubleand single-strand DNA breaks which disrupt cell proliferation. These aberrant DNA structures induce signaling pathways involved in the DNA damage response (DDR), which regulate these cell cycle checkpoints and DNA repair. Two key kinases involved in this response are ataxia telangiectasia-mutated (ATM), activated primarily by DNA double-strand breaks, and ATM- and Rad3-related (ATR), activated by a broad spectrum of DNA lesions. ATM and ATR work in a coordinated fashion to effect homologous recombination (HR) repair, involving a gradual switch from ATM to ATR activation [25]. This is facilitated by two specific substrates of ATM and ATR, CHK2 and CHK1, respectively. Activation of the G1/S checkpoint by DNA double-strand breaks is regulated by p53, which is a direct substrate of ATM/ATR, as well as CHK1 and CHK2 [26-28]. Activation of the G2/M checkpoint by DNA double-strand breaks is more selectively regulated by the ATR-CHK1 [29,30].
Several inhibitors of CHK1 and CHK2 are being investigated as radiosensitizers, both in pre-clinical and clinical settings. The DDR in tumor cells differs significantly from that in normal cells, with tumor cells often having defective DNA damage signaling through loss of ATM or p53 mutations. Since p53 mutations compromise efficient G1 checkpoint signaling, the DDR in tumor cells carrying these mutations depends more heavily on the ATR-CHK1-activated G2/M checkpoint for cell cycle arrest. Thus, checkpoint inhibitors selectively targeting the ATR-CHK1 pathway have the potential to sensitize tumor cells to radiation-induced DNA damage without sensitizing normal cells with wild type p53. Such compounds are also being investigated in this context and appear to be potent and selective radiosensitizers of cancer cells [31,32].
CHK1 functions in HR repair, stabilizing replication forks, and inhibiting apoptosis. Many CHK1 inhibitors exhibit radiosensitization in clinical models [33,34]. For example, MK-8776, a potent and selective ATP-competitive CHK1 inhibitor that increases cell sensitivity to DNA-damaging agents, is undergoing phase II clinical trials with cytotoxic chemotherapy (e.g., gemcitabine, cytarabine, etc.). VE-821, a potent ATR inhibitor, has been shown to enhance the radiosensitivity of pancreatic cancer cells both in vitro and in vivo [35]. As expected, this inhibitor decreased HR repair. Both CHK1 and ATR inhibitors exhibit their radiation enhancement regardless of the mutational status of p53, which is consistent with the loss of checkpoint control at both the G1/S and G2/M checkpoints as well as compromised HR repair.
Many human tumors exhibit overexpression of WEE1, which selectively regulates the G2/M checkpoint in response to DNA damage by inhibiting CDK1. Pre-clinical studies have shown that WEE1 also participates in DNA repair by stabilizing DNA replication forks. WEE1 inhibitors, therefore, have the potential to remove the G2/M block and to compromise WEE1-mediated DNA repair. MK-1775, a selective, small molecule inhibitor of WEE1, has been shown to radiosensitize multiple human cancer cell lines [36] and is currently being investigated in a clinical trial in conjunction with fractionated RT in patients with glioblastoma multiforme [37]. Radiation enhancement by WEE1 inhibitors is dependent on the p53 status in cell culture studies, where mutated p53 cells are preferentially radiosensitized whereas wild type p53 cells show no significant radiation enhancement [38]. Cells in G2 phase are preferentially sensitized by WEE1 inhibitors, reflecting the loss of WEE1-mediated DNA repair.
Immune Modulators and Radiotherapy
Ever since the early 1960s, when presence of tumor specific antigens was convincingly demonstrated [39,40], many attempts have been made to exploit the power of host immunity against these tumor antigens. Successful clinical results have been elusive. Only within the past 7 years, with the advent and success of immune check point inhibitors as a treatment for advanced metastatic cancers, has immunotherapy moved to the center stage of clinical oncology [41-44]. Further, excitement was generated when local radiotherapy was combined with check point inhibitors and widespread metastatic tumor clearance was achieved [45-47]. In the past 2 years, numerous clinical trials have been initiated combining either fractionated radiotherapy or stereotactic hypofractionated radiotherapy with check point inhibitors [48,49]. Pre-clinical immuno-radiobiology research is underway to address some key pressing questions related to the optimization of radiation dose, timing, and fractionation schedules of RT for combination therapy with these inhibitors [49,50].
1. Immune check point inhibitor and radiotherapy
CTLA4 is a CD28 homolog that acts the early stages of immune response by affecting the interaction between T cells and antigen-presenting cells in the lymph nodes. PD-1/PDL1 suppress T cells later on during the effector phase of the immune response, especially in the periphery. Anti-CTLA4 therapies increase T cell activation by enhancing proliferation and reducing Treg immunosuppression. On the other hand, anti-PD-1/PD-L1 therapies act to restore T cells that are exhausted due to chronic exposure to carcinogen and the accumulation of mutations over time [44]. There are several new classes of agents that are able to inhibit immune check points. Check point inhibitors targeting CTLA4 (ipilimumab), PD-1 (nivolumab, pembrolizumab) and PD-L1 (atezolizumab and durvalumab) have been developed with demonstrated efficacy in unresectable or metastatic refractory melanoma, and advanced squamous and non-small cell lung cancers and are being developed for many other tumors as well (Fig. 2).
Like many other therapeutic regimen used in combination with RT (e.g., cytotoxic chemotherapy, targeted therapy), key radiobiological parameters play important roles when combined with immune checkpoint inhibitors [51]. Clinical and pre-clinical studies are underway to address some unknowns such as the effect of radiation dose per fraction, irradiated tumor volume and time sequence of the drug administration. Hypofractionation with 8 Gy appears to be more effective than lower dose per fraction irradiation in generating so called ‘abscopal’ effect (24 Gy in 3 fractions yielded a larger effect than 30 Gy in 5 fractions) [48]. In the pre-clinical setting, abscopal effects have been reported using these dose regimens in combination with CTLA4 blockade [48]. The other factor is the timing and sequence of the inhibitor administration relative to RT. At present, administration of the inhibitor prior to RT is preferred because the high single dose of hypofractionated RT may destroy tumor vasculature thereby preventing the cytotoxic T cell from reaching the core of large solid tumors. However the optimum timing remains an area of investigation since lymphocytes are among the most radiosensitive cells of body and the damaging effects of prior radiation on a subsequent immunological event based on T lymphocytes needs to be better understood. Although the combined effects of RT and immune checkpoint inhibitor on the abscopal response is exciting, it is still considered a rare event [49]. Many other immune suppressive factors in the tumor microenvironment are in play in addition to the immune checkpoints [50].
2. Immune suppressors and radiotherapy
The tumor microenvironment harbors several immune suppressors such as myeloid derived macrophages, regulatory T cells, and TGFβ, to name a few [51]. The local tumor microenvironment is usually characterized as being acidic, having low oxygen and deprived of micronutrients. These conditions favor attracting myeloid derived macrophages and activating them to so-called M2 type macrophages, also known as tumor associated macrophage (TAM). TAMs are anti-inflammatory and immune-regulatory, and are therefore tumor promoting, whereas activated M1 type macrophages are pro-inflammatory and tumoricidal. TAM infiltrations have been shown to have a negative prognostic relevance for most tumor types [52]. Thus several attempts have been made to target these cells. Among them, the colony stimulating factor 1 receptor (CSF1-R) axis has gained the most attention and a CSF1-R inhibitor is undergoing a clinical trial as a monotherapy [53]. Since activated macrophages play a significant role in the pathogenesis of radiation induced normal tissue damage, we and others have studied the effect of suppressing macrophage mediated pro-inflammatory cytokines on the normal tissue injury (e.g., skin and CNS). Suppression of pan-cytokines have significantly mitigated the radiation induced skin damage and cognitive dysfunction in the brain [54,55]. Recently, Acharya et al. [56] have published results of a similar abrogation of the radiation induced cognitive dysfunction by depleting activated microglia using a CSF1-R inhibitor.
In this context, it would be highly desirable to determine whether the depletion of TAM with CSF1-R inhibitor would enhance the tumor radiation response or not. If it shows an enhancing effect, this approach would be another novel strategy for enhancing the therapeutic ratio. Furthermore, a combination with immune checkpoint inhibitors and the depletion of TAM with CSF1-R inhibitors would be a potentially exciting new approach.
TGFβ is a multifunctional and pleiotropic cytokine affecting many cellular processes including epithelial cell growth, mesenchymal cell proliferation, and extracellular matrix production. Irradiation, even at low doses, is one of the few exogenous factors known to induce TGFβ activation and TGFβ is believed to play a central role in mediating radiationinduced anti-tumor immunity [57] and tissue fibrosis (skin, lung) [58,59]. In advanced cancers, TGFβ promotes tumor growth and metastasis [60]. Using an anti-TGFβ monoclonal antibody, phase I clinical trials were carried out in patients with advanced malignant melanoma and renal cell carcinoma [61]. A pre-clinical study was also carried out with combining radiotherapy with an anti-TGFβ agent. Radiosensitivity of tumor cells were enhanced both in vitro and in vivo [62]. Recently, a phase I clinical study was conducted in metastatic breast cancer patients using a TGFβ blockade and focal hypofractionated RT administered to the metastatic foci. The data suggest that the combined approach induces an abscopal response in a few patients [63]. Blocking an immunosuppressive cytokine alone with local RT may not be sufficient, but multi-pronged approach such as immune check point inhibitors and activated M2 macrophage depletion need to be studied further, because they may prove to be a successful approach.
The use of immunosupressive approaches other than M2 macrophages, such as myeloid-derived-suppressor cells, MDSC, is worth considering since these immunosuppressive myeloidderived cells also infiltrate tumors after RT. For example, STING-dependent MDSC infiltrate tumors after radiation exposure providing an immunosuppressive environment and their blockade enhances the effectiveness of RT [64]. Regulatory T-cell depletion has also been shown to enhance RT efficacy in pre-clinical lung and colon cancer models [65].
Strategies to Prevent/Mitigate/Treat the Normal Tissue Injury
Recent advances in our understanding of the cellular and molecular pathways leading to tissue and organ damage have provided a novel insight into the mechanisms of pathogenesis. Strategies aimed at reducing or counteracting oxidative stress and the resulting excessive production of reactive oxygen and nitrogen chemical species (ROS and RNS, respectively) and pro-inflammatory cytokines have been covered in detail in this journal [66]. In this section, we will briefly review new emerging strategies involving interference of the pathogenic cell signaling pathways and role of radiation-induced senescence.
1. Inhibition of mTOR and HIF pathway
We have discussed the role of rapamycin and its analogs as a radiosensitizer in the foregoing section on the PI3K/mTOR pathway. The radiosensitizing effect was shown in mostly tumor tissues. Interestingly, similar inhibitors have been shown to protect or mitigate the radiation induced normal tissue injury [67,68]. The underlying mechanisms for these mitigating effect are not well understood, but experimental correlative data suggest that the mitigation may be mediated through anti-apoptosis, anti-senescence and less γ-H2AX, etc. It is interesting to note that rapamycin slows aging and extends life span in a variety of species from worm to mammals. In this context, metformin, another radiosensitizer, included in our previous discussion, demonstrates radioprotection in some normal tissues, presumably through AMPK upregulation leading to mTOR inhibition (Fig. 1).
Inhibition of the HIF-1α pathway has been shown to mitigate radiation-induced gastrointestinal toxicity [69,70]. Using a small molecule inhibitor of prolyl hydroxylases, the acute gastrointestinal toxicities following high dose of irradiation of the abdomen significantly mitigated its acute and late toxicity. The inhibitor increases HIF-2 expression, improves epithelial integrity, reduces apoptosis and increases intestinal angiogenesis [69]. Interestingly, the inhibitor did not mitigate against the bone marrow syndrome following total body irradiation.
2. Role of senolytics
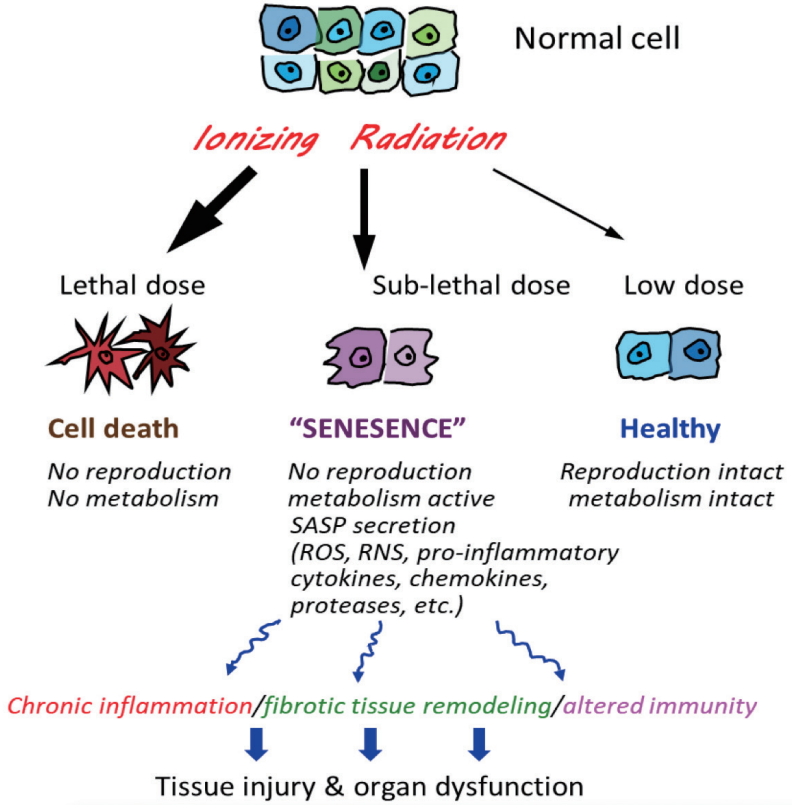
Cellular senescence, which is a normal consequence of aging, can result from DNA damage such as that found after radiation exposure as well as oxidative stress, and chronic inflammation. Senescent cells lose the proliferative potential normally found in replication-competent cells and becomes resistant to apoptosis, with an increase in metabolic activity. These changes are often accompanied by the development of a phenomenon known as senescence-associated secretory phenotype (SASP) (Fig. 3). The SASP entails release of proinflammatory cytokines and chemokines, tissue-damaging proteases, factors that can affect stem and progenitor cell function, homeostatic factors, and growth factors, among others. Hence, senescent cells that express the SASP can have substantial local and systemic pathogenic effects.
Laboratory studies have confirmed the importance of senescence as a cause of radiation toxicity in bone marrow, skin and lung and of toxicity from DNA damaging chemotherapy [1,71-74]. Indeed, a recent study in the radiation biology literature showed that radiation-induced lung fibrosis in mice was reversed completely following treatment with ABT-263, a known senolytic drug targeting Bcl2/xL [75]. Both epithelial cell and endothelial cell populations have been implicated [76]. Using another senolytic (e.g., HSP-90 inhibitor), a significant mitigation of radiation-induced skin injury was found in mice, especially late injury endpoints such as leg contraction and fibrosis [77].
Multiple pharmacological strategies are under investigation to remove senescent cells from non-genetically modified animals, using so-called ‘senolytic agents’, including small molecules, peptides, and antibodies with varying degrees of success [78,79]. Some senolytic agents are cell and tissue specific; others are not. Several pathways have been identified including the Bcl-2 family, PI3K/mTOR, tyrosine kinase receptors, and HSP-90, to name a few. It is interesting to note that the most senolytic drugs were initially being developed as anti-cancer agents, since some of the signaling pathways in tumor and senescent cells overlap each other. In the near future, more pre-clinical radiobiological studies are needed to determine the role of senolytics as either mitigators or therapeutics in normal tissue and organ injury but also therapeutics to enhance tumor response.
Conclusion
Recent advances in the cellular and molecular pathways of tumor growth and the pathogenesis of tissue and organ injury provide novel strategies to enhance the radiation therapeutic ratio. Radiobiological research is aimed to optimize combined therapeutic regimens of radiation and targeted therapies and immunotherapeutics. Efforts to minimize or mitigate the risk of radiation injury constitute a promising direction of future study.